Introduction
Corneal dystrophies are rare hereditary corneal diseases that are usually bilateral, symmetric, slowly progressing, and are unrelated to systemic factors [1]. The disease is manifested in the form of the deposit accumulation in the various layers of the cornea. Previously, the classification of corneal dystrophy had been mainly based on the location of corneal opacity and the characteristics of the phenotype. Recently, the International Committee for Classification of Corneal Dystrophy (IC3D) developed a new classification system based on different layers of the cornea of which that is affected [2,3]. Corneal dystrophies are currently divided into epithelial and subepithelial dystrophies, epithelial-stromal transforming growth factor β induced (TGFBI) dystrophies, stromal dystrophies, and endothelial dystrophies.
Granular corneal dystrophy type 2 (GCD2) was first described by BĂźcklers in 1938, which, however, was considered as a mild variant of GCD1 for a long period of time [2]. GCD2 is inherited by autosomal dominant pattern with very high penetrance, showing granular deposits, linear lesions in deep stroma and superficial diffuse haze in advanced stage [2,4]. Because this corneal dystrophy showed different phenotype compared to GCD1, it was separated from GCD1 in 1992. As a pedigree of the dystrophy was established with a family originated from Avellino district of Italy, it was named Avellino corneal dystrophy at first [5]. Then, in 2008, IC3D recommended expressing this corneal dystrophy as GCD2 [3].
This review aims to outline key knowledges regarding genetics, epidemiology, clinical features, and management of GCD2.
Genetics, Epidemiology, and Pathophysiology
GCD2 is caused by mutations of TGFBI gene on chromosome 5q31 and is inherited by autosomal dominant fashion with high penetrance. One of the most common mutations is p.Arg124His mutation [6,7]. The other mutations inducing morphologic or histologic characteristics similar to that of GCD2 need further evaluation [8-11].
Even though GCD2 was once referred as Avellino corneal dystrophy, named after the Italian district as mentioned above, its prevalence is actually higher in East Asian countries such as Korea and Japan [12], being about 11.5 affected persons per 10,000 individuals in Korea [13]. GCD2 accounts for 72% to 91% of TGFBI-related corneal dystrophies in Korea and Japan, 67% in China, while it accounts for 36% in the United States and 3% in Poland [14,15].
TGFBI encodes for an extracellular matrix protein (TGF-BI protein [TGFBIp], also called keratoepithelin) involved in cell adhesion, migration, and proliferation [16]. Mutated TGFBIp aggregates and results in the formation of abnormal insoluble deposits within the corneal stroma. Recent research also showed that mutated TGFBIp itself affects the corneal fibroblast so that the function and characteristics of corneal fibroblasts would be changed in GCD2 [16,17].
Clinical Features
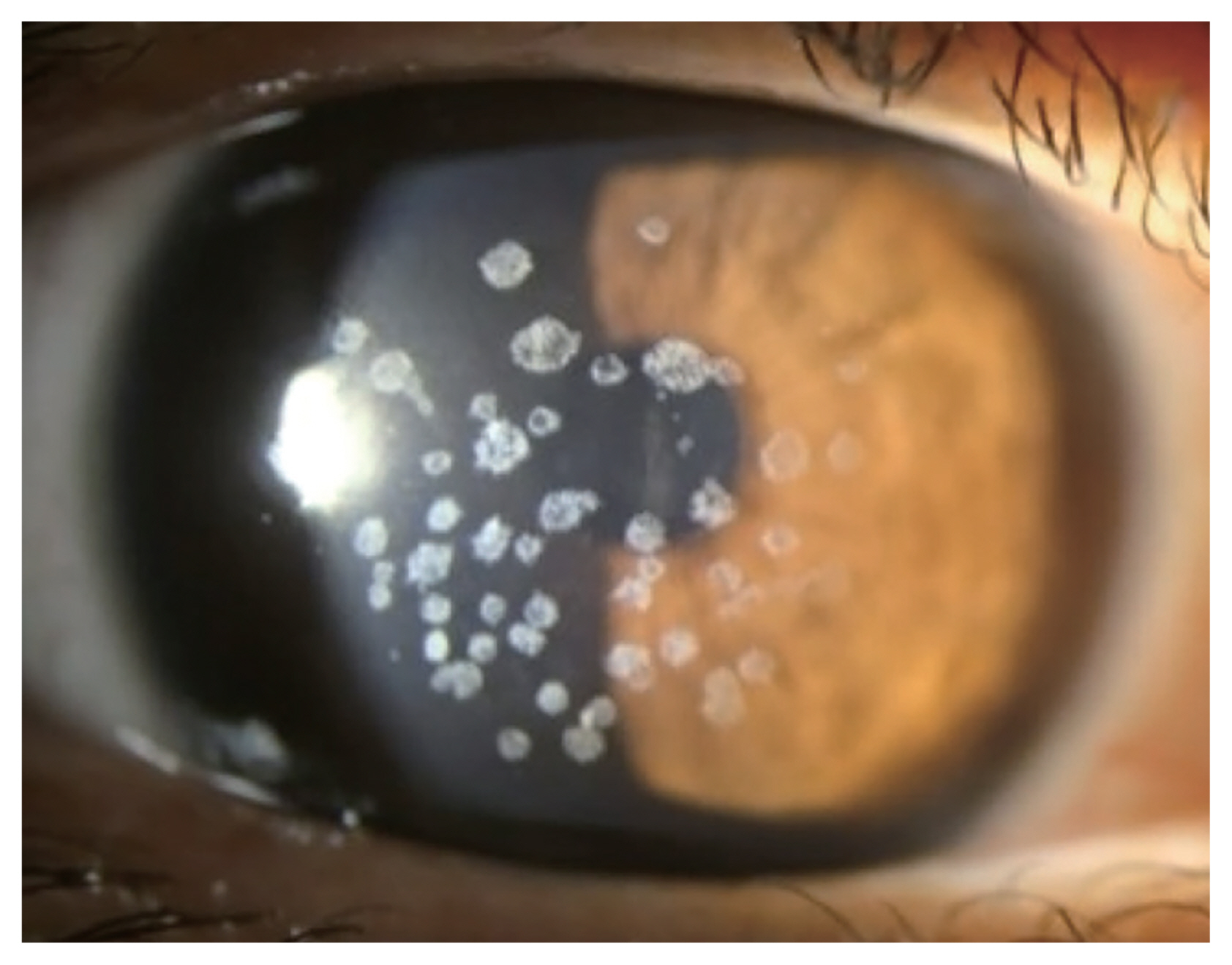
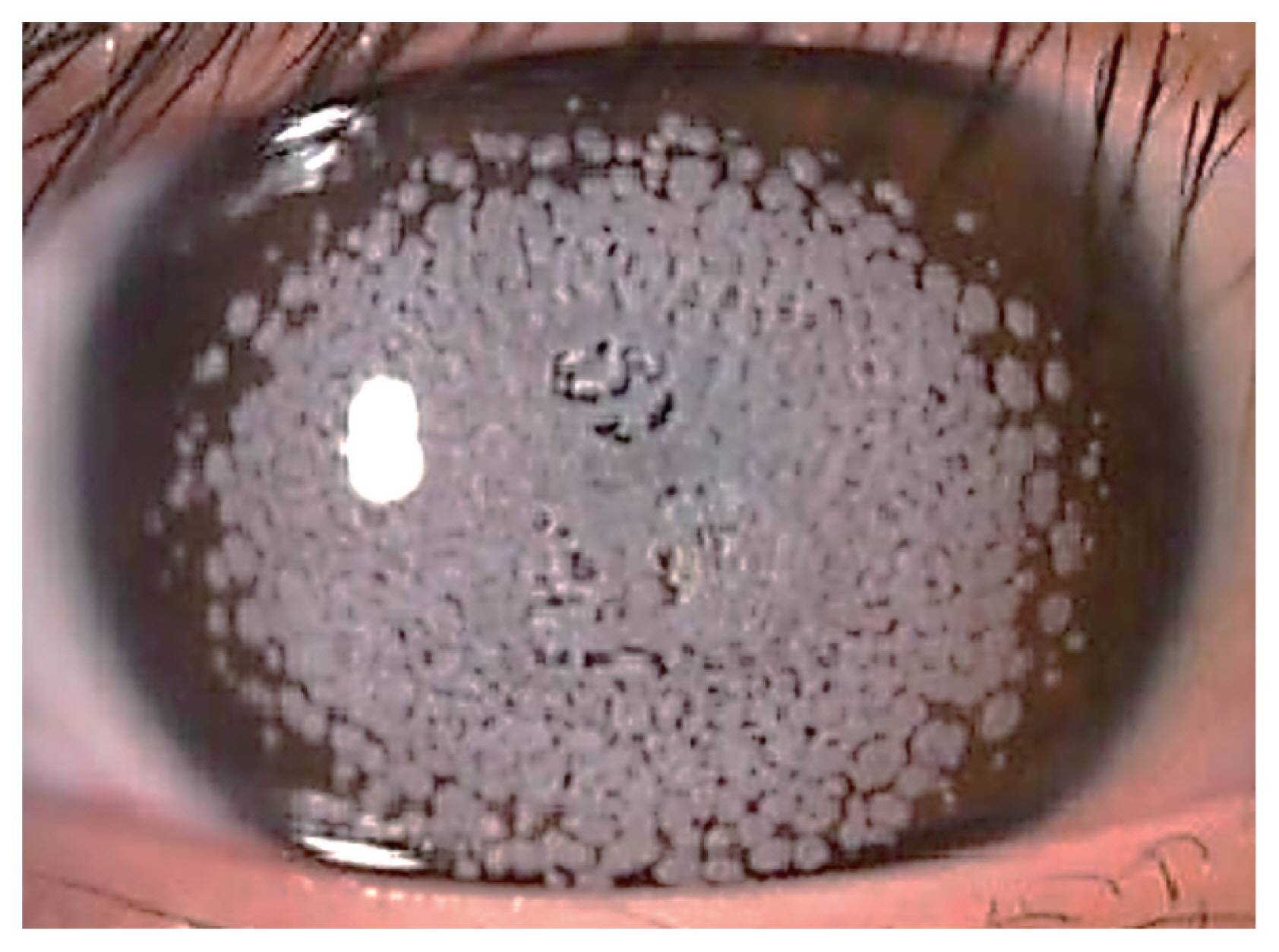
Small gray to white, well-circumscribed deposits appear in superficial stroma initially, and become larger into granular shapes (Fig. 1). As the disease progresses, the deposits become even larger and more pronounced. The center of the deposits can be dropped out, and extruded deposits cause corneal erosions, resulting in pain, photophobia, discomfort, and foreign body sensation [18]. However, recurrent corneal erosion are less frequent compared to GCD1 [12]. Center of the deposit forms a punched-out lesion leaving the lesion ring or discoid shaped with transparent centers. GCD2 also can show diffuse haze in anterior stroma with aging, reducing visual acuity.
Shapes of corneal opacities differ in stromal layers, yet endothelium is spared. In anterior to mid stroma, it tends to be stellate and spiked, partially translucent in retroillumination while more linear and lattice like deposits are found in deeper stroma. Linear deposits are whiter and opaque, less refractile than that of lattice corneal dystrophy type 1.
Granular lesions are composed of hyaline, where linear lesions are composed of amyloid and linear deposits characteristically intersect. The granular opacities are shown as eosinophilic deposits using light microscopy under the Masson trichrome staining [19]. Upon transmission electron microscopy, rod-shaped electron dense deposits are found in anterior stroma. GCD2 heterozygous keratocytes are enlarged compared to normal corneal keratocytes, enabling visualization of intracellular organelle [20].
Clinical manifestations differ depending on homozygosity; homozygous GCD2 tends to be diagnosed earlier, be more severe and progresses more rapidly with significant visual impairment at early ages (Fig. 2). Homozygous patients can be diagnosed as early as at the age of 3 years, when white deposits on cornea becomes apparent [11]. Heterozygous patients are usually diagnosed as teenagers or young adults, although earliest can be at the age of 8 years [11]. Severity of heterozygote GCD2 can vary among the patients [9,10,21-30]. Several severe phenotypic forms were found to be caused by different types of compound heterozygous mutation [31].
Opacities are usually found around the center of the cornea, sparing periphery near the limbus. A study done by Lee et al. [32] showed that corneal neovascularization affects this feature. For instance, a GCD2 patient undergoing phthisis with corneal neovascularization had no deposits on the corneal periphery. In a GCD2 heterozygous patient with pterygium, opacities are located further away from limbus on the side of cornea where pterygium is present due to neovascularization, compared to the opposite side of the cornea without pterygium, where opacities are present closer to the limbus. Also, the opacities near pterygium tend to be less distinctive, implying resorption of the deposits. These findings show that characteristic of corneal vascular supply may be related to clearing of GCD2 deposits. Another supporting case is that clear incision near limbus during cataract surgery does not aggravate the disease whereas refractive surgeries such as laser in situ keratomileusis (LASIK) and laser epithelial keratomileusis (LASEK) performed at the central part of cornea, which is located far from the vascularized limbus, usually result in exacerbation of GCD2 [33].
Homozygous GCD2, compared to heterozygotes, shows more aggressive presentation and progression in terms of number of opacities, growth in size and fusion of the deposits. Moreover, patientâs vision is impaired more rapidly that he or she requires surgical treatments such as phototherapeutic keratectomy (PTK) or corneal transplantation even at childhood and recurrence is more frequent and faster [11]. Range of peripheral cornea that is clear of opacities decreases with age. In homozygous GCD2, the confluent opacities appear early but deep linear deposits do not form unlike heterozygous GCD2. Homozygous GCD2 can show two distinct phenotypes, type I opacity shows spot like gray-to-whitish deposits [11,34,35] and type II is characterized by grayish reticular shaped opacities with intervening translucent spaces resulting in better best corrected vision than type I opacity [36,37].
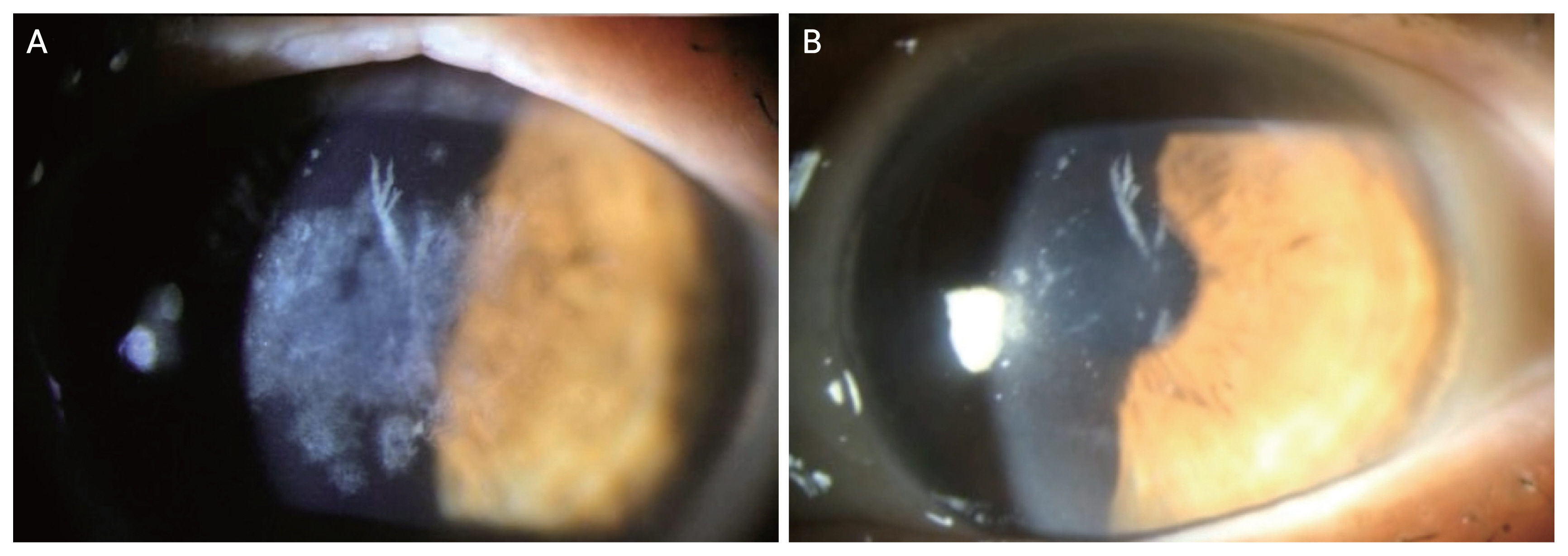
Several studies examined exacerbation or recurrence of GCD2 following different refractive surgeries such as LASIK, LASEK, and photorefractive keratectomy (PRK) [38-43]. After an uncomplicated LASIK surgery, opacities tend to get worse, forming multiple small granular deposits in between the flap and the stromal bed (Fig. 3), whereas fine deposits form at superficial stroma following LASEK and PRK. Exacerbation is generally more severe and final visual acuity is worse after LASIK compared to post-PRK status [42,43]. There is also a recent report where GCD2 aggravated after small incision lenticule extraction (SMILE) procedure [44].
Management
Conservative managements, such as artificial tears or therapeutic contact lenses are utilized when GCD2 causes recurrent corneal erosions. Further surgical managements are considered when patients experience decrease in visual acuity, contrast sensitivity and therefore deterioration of functional vision. Choice of surgical method depends on depth and extent of the deposits and final treatment goal is to remove the opacities and postpone keratoplasty as late as possible.
PTK aims to remove opacities located in anterior stroma (Fig. 4A, 4B). It is easily applicable and does not impose risk of graft rejection. It delays timing of keratoplasty such as deep anterior lamellar keratoplasty (DALK) or penetrating keratoplasty (PKP) and can be done repeatedly despite recurrence and postkeratoplasty status [45]. Along with preoperative slit-lamp examination, Fourier domain optical coherence tomography (FD-OCT) can be utilized in order to visualize the depth of the corneal deposits, enable the surgeon to plan and minimize the ablation depth of PTK for removal of the deposits [46,47].
Recurrence rate reported in various studies range from 0% to 100% due to different definitions of recurrence and range of follow-up durations [48-54]. Deposits relapsed faster in homozygous patients at 18 months after first PTK treatment, and at 3 months following second or third treatment [11]. In a study done by Inoue et al. [55], GCD2 recurred after 9.5 months in four homozygous patientsâ eyes, compared to 38.4 months in seven heterozygous GCD2 eyes. The use of mitomycin C (MMC) during PTK has been tried in some clinics. A 3-year follow-up after surface ablation using MMC showed that GCD2 still aggravated afterwards [56]. Use of MMC is not recommended as MMC would induce apoptosis of keratocyte which would do the work of reabsorption and degradation of the TGF-BIp located in the stroma [57,58].
PTK can be also done prior to or after different ocular surgeries. For example, PTK can be applied to cornea under progression after LASIK, with better effect when the LASIK flap is removed [54]. When a patient requires both cataract surgery and PTK, PTK should be done prior to, then different formulas for calculation of intraocular lens power after PTK can be used [59-61].
Keratoplasty such as anterior lamellar keratoplasty (ALKP), DALK, and PKP can be considered for deep stromal deposits and when PTK is no longer repeatable due to excessive corneal thinning, astigmatism, or scarring. Since corneal endothelium is spared in GCD2, ALKP and DALK is preferred over PKP. Recurrence rates are reported ranging from 0% to 71% depending on length of follow-up period, and some studies also did not mention subtypes of GCD [62-65].
Since corneal epithelial cells are suggested to be the origin of TGFBIp resulting in corneal deposits of GCDs in previous studies, limbal cell transplantation can be used. However, concerning the rejection of the transplanted limbal cells, it is not yet practically used [66-69]. Another possible treatment method reported for the removal of corneal deposits is corneal electrolysis. In a study done by Mashima et al. [70], corneal electrolysis was applied to two homozygous eyes that recurred following keratoplasty, where it recurred with fine granular deposits after three years. Long-term clinical outcome is not yet studied.
Future Perspectives
Further studies are needed to evaluate long-term clinical outcomes of different treatment modalities. Also, different management approaches other than surgical or interventional methods can also be considered for treatment of GCD2. For instance, studies have found that lithium chloride decreases TGFBI protein production [71] and combined treatment of melatonin and rapamycin inhibits TGF-BI protein expression while increasing degradation of the molecule [17,72,73]. In addition to pharmacological agents, gene therapy such as small interfering RNA (siRNA), short hairpin siRNA may be used to silence mutant TGFBI expression [74,75]. Genome editing technique such as CRISPR/Cas9 may be used to target genes site specifically. However, one of the problems of using gene therapy technique is the unwanted off-target effects, which may affect or silence the opposite normal allele or other genes [31,76]. Developing safe delivery methods free from unwanted off-target effects would be essential for specifically targeted gene therapy for GCD2.
Conclusion
GCD2 is a stromal corneal dystrophy inherited by an autosomal dominant manner. The dystrophy is characterized by well-circumscribed granular opacities, deep linear lesions and diffuse corneal haze resulting in decrease in visual acuity. Circumscribed superficial granular opacities may sometimes become punched-out lesions causing pain due to corneal erosions. Severity and pace of progression vary depending on the genotype, homozygotes being the most severe. Current managements include conservative treatment such as artificial tears and therapeutic contacts lenses for corneal erosions, and surgical interventions depending on the depth of the stromal deposits: PTK for anterior opacities and keratoplasty (DALK) for deeply located deposits. Further studies can be considered to develop different treatment modalities such as pharmacological agents or genetic therapy, providing wider range of options for GCD2 patientsâ life-long management.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print