Ischemia-reperfusion injury (I/R injury) is correlated with certain unique pathological outcomes in which apoptotic cell death plays a central role (such as myocardial infarction and peripheral vascular disease). Apoptosis causes failure of contractile recovery after heart muscle injury [1]. Hypoxic and sudden oxidative stresses cause a rapid increase in oxygen-free radicals and an overload of intracellular calcium that may directly contribute to this type of tissue death [2]. We were particularly interested in I/R injury-mediated apoptosis in the retina because retinal vein occlusion is one of the most common retinal vascular diseases [3,4]. Retinal degeneration induced in rats by the transient elevation of intraocular pressure shows an extensive loss of the neuronal elements of the retinal ganglion cell layer and the inner nuclear layer [5]. Once retinal degeneration progresses to neovascular glaucoma, there are few options for the treatment of retinal vein occlusion.
Crystallins, well-known constitutive proteins of mammalian lenses [6], have recently been identified as having an anti-apoptotic role. In particular, αA- and αB-crystallins act as decoy molecules to block caspase 3 and caspase 6. A mechanism similar to the apoptotic inhibition by crystallin was demonstrated with the pro-apoptotic factors Bax and Bcl-Xs [7]. The overexpressed αB-crystallin blocks the formation of reactive oxygen species [1]. During ischemic preconditioning, the bovine B2 chain of α-crystallin is activated through the phosphorylation of the serine residues (Ser-45 and Ser-59) mediated by ERK1/2 and p38, respectively, thereby acting as an effector protein [8-12]. More recently, post-translational modifications of mouse αA-crystallin have been reported [13,14]. The N-terminal acetylation and the variable C-terminal truncation of αA-crystallin are regarded as important modifications for the activation of crystallin proteins [15].
The vitreous body, a comparatively acellular tissue, consists primarily of water, collagen, and hyaluronic acid [16]. The vitreous body is surrounded primarily by the neurosensory retina but also by the ciliary body, iris, and lens. Large molecules such as proteins may accumulate in the vitreous body due to local secretion [17], through filtration from the blood [18], or by diffusion from the surrounding tissues and vasculature. The simplicity of the vitreous body is an advantage for the study of proteomics in ophthalmology. We examined the relationship between the crystallins of the vitreous body and an I/R injury-related retinal eye disease model in male Sprague-Dawley (SD) rats.
Materials and Methods
Animals
Twenty-four SD rat (6 to 8 weeks old, 150 to 200 g) were procured from Orient Bio (Seongnam, Korea) and were accommodated under specific pathogen-free conditions. All trials were performed in compliance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. The four groups were divided as follows: 1) control group, 2) 24 hours after I/R injury, 3) 48 hours after I/R injury, and 4) 72 hours after I/R injury. Control rats received an injection of the same volume of saline on the same schedule.
Induction of ischemia-reperfusion injury
The retinal artery was clamped for induction of a hypoxic ischemia-reperfusion retinal injury and then released 30 minutes later. The blood vessel was microscopically monitored to ensure the blockage and release of the blood flow.
Sample preparation
A ketamine (15 mg/kg) and xylazine (85 mg/kg) mixture was injected into the abdominal cavity of each rat, and the vitreous bodies were obtained. The vitreous bodies were washed twice with a Tris-sorbitol buffer (pH 7.0) and solubilized with 1 mL of lysis buffer containing 8 M urea (Sigma, St. Louis, MO, USA), 2% (w/v) CHAPS (Sigma), 50 mM DTT (Sigma), 40 mM Tris (Sigma), 2% (v/v) pH 3 to 10 or pH 5 to 8 carrier ampholytes (Bio-Rad, Hercules, CA, USA), and a trace of bromophenol blue. For the solubilization of proteins, tip-probe sonication was used for 3 × 10 seconds (total 30 seconds) on ice. The quantity of protein was analyzed using Bradford reagent (Sigma).
Two-dimensional electrophoresis
Two-dimensional electrophoresis (2-DE) was performed five times using the Amersham Biosciences system. One milligram of whole vitreous lysate was applied to each immobilized pH gradient (IPG) strip (Bio-Rad, pH 3 to 10 NL or pH 5 to 8 strip), which had been rehydrated in 9 M urea, 2% CHAPS, 50 mM DTT, 0.2% Bio-Lyte pH 3 to 10 or pH 5 to 8 carrier ampholyte, and 0.001% bromophenol blue (Bio-Rad, ReadyPrep 2-D start kit rehydration/sample buffer). Pre-isoelectric focusing (pre-IEF) and isoelectric focusing (IEF) were performed using pre-made 18 cm IPG strips on a Multiphor II electrophoresis system (Amersham Biosciences, Piscataway, NJ, USA). The pre-IEF was performed linearly up to 500 V for 1 hour and subsequently held at 500 V for 2 hours. The IEF was then performed with a linear increase up to 3,500 V over 3 hours and then held at 3,500 V for 60,000 Vh. For the second dimension, the IPG strips were equilibrated in equilibration buffer I (375 mM Tris-Cl, pH 8.8, 20% glycerol, 2% SDS, and 6 M urea containing 2% dithiothreitol [DTT]) for 30 minutes then moved for [DTT]) to equilibration buffer II (375 mM Tris-Cl [pH 8.8], 20% glycerol, 2% SDS, and 6 M urea containing 2.5% iodoacetamide) for 1 hour on a rocker. The strips were subjected to electrophoresis on a 15% SDS-PAGE using a Protean Plus Dodeca Cell (Bio-Rad). The protein marker used was a pre-stained protein ladder (Fermentas 11 to 170 kDa). Gels were stained with Coomassie Blue G250 (Sigma).
The normalization of proteins was automatically calculated by a computer program. The ImageMaster 2D Platinum Software (ver. 5.0, Amersham Bioscience) calculated the relative volume (%vol) for each spot. These relative volumes are normalized values that remain relatively independent of irrelevant variations between gels caused by varying experimental conditions.
In-gel digestion electrophoresis
The excised spots were cut into smaller pieces and digested using trypsin (Promega, Madison, WI, USA) as previously descrbed [19-21]. For the matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF/MS) analysis, the trypsin peptides were concentrated by poros R2 and oligo R3 columns (Applied Biosystems, Foster City, CA, USA) and then eluted in α-cyano-4-hydroxycinnamic acid [20]. The spectra were obtained using a Voyager DE PRO MALDI-TOF spectrophotometer (Applied Biosystems). The protein database searches were performed by Matrix Science (http://www.matrixscience.com) using monoisotopic peaks. The protein function was cited either from the ExPASy database (http://kr.expasy.org), the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov), the Kyoto Encyclopedia of Genes and Genomes (http://www.genome.ad.jp/kegg), or from the publications cited below.
Western blotting of crystallins and detection of phosphorylation
The samples were electrophoresed on a 15% polyacrylamide gel and transferred to a nitrocellulose membrane (Bio-Rad). The membranes were blocked for 2 hours in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) buffer (Sigma) containing 5% skim milk (BD Biosciences). They were then incubated with anti αA crystallin polyclonal antibody (Stressgen, Victoria, BC, Canada), anti-αB crystallin polyclonal antibody (Stressgen), anti-β crystallin monoclonal antibody (Stressgen), anti-phospho αB crystallin (Ser-19) polyclonal antibody (Stressgen), anti-phospho αB crystallin (Ser-45) polyclonal antibody (Stressgen), anti-phospho αB crystallin (Ser-59) polyclonal antibody (Stressgen), anti-ERK1/2 antibody, and anti-phosphorylated ERK1/2 (pERK1/2) antibody (Invitrogen).
After being washed three times in TBS-T, the membranes were incubated with anti-rabbit IgG secondary antibody or anti-mouse IgG secondary antibody at a dilution of 1 : 2,000 in TBS-T containing 1% skim milk for 1 hour at room temperature. The final products were visualized using a BCL kit (Amersham Biosciences).
Results
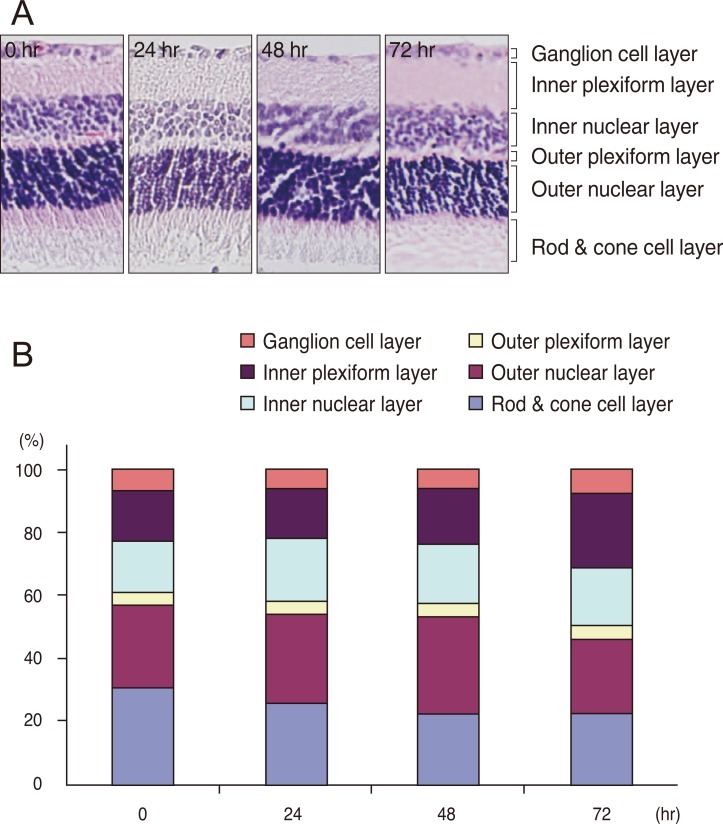
Histopathologic changes in retinal elements
The retinas of the four groups of rats were observed after the induction of I/R injury. Fig. 1 shows that the ganglion cell layer morphology was changed at 24 hours and 48 hours after I/R injury. The inner nuclear layer and the outer nuclear layer were thick at 24 hours and 48 hours after I/R injury. The ganglion cell layer, the inner nuclear layer and the outer nuclear layer were recovered at 72 hours after I/R injury. However, the rod and cone cell layer was not recovered at 72 hours after I/R injury (Fig. 1B). These results support the hypothesis of I/R injury.
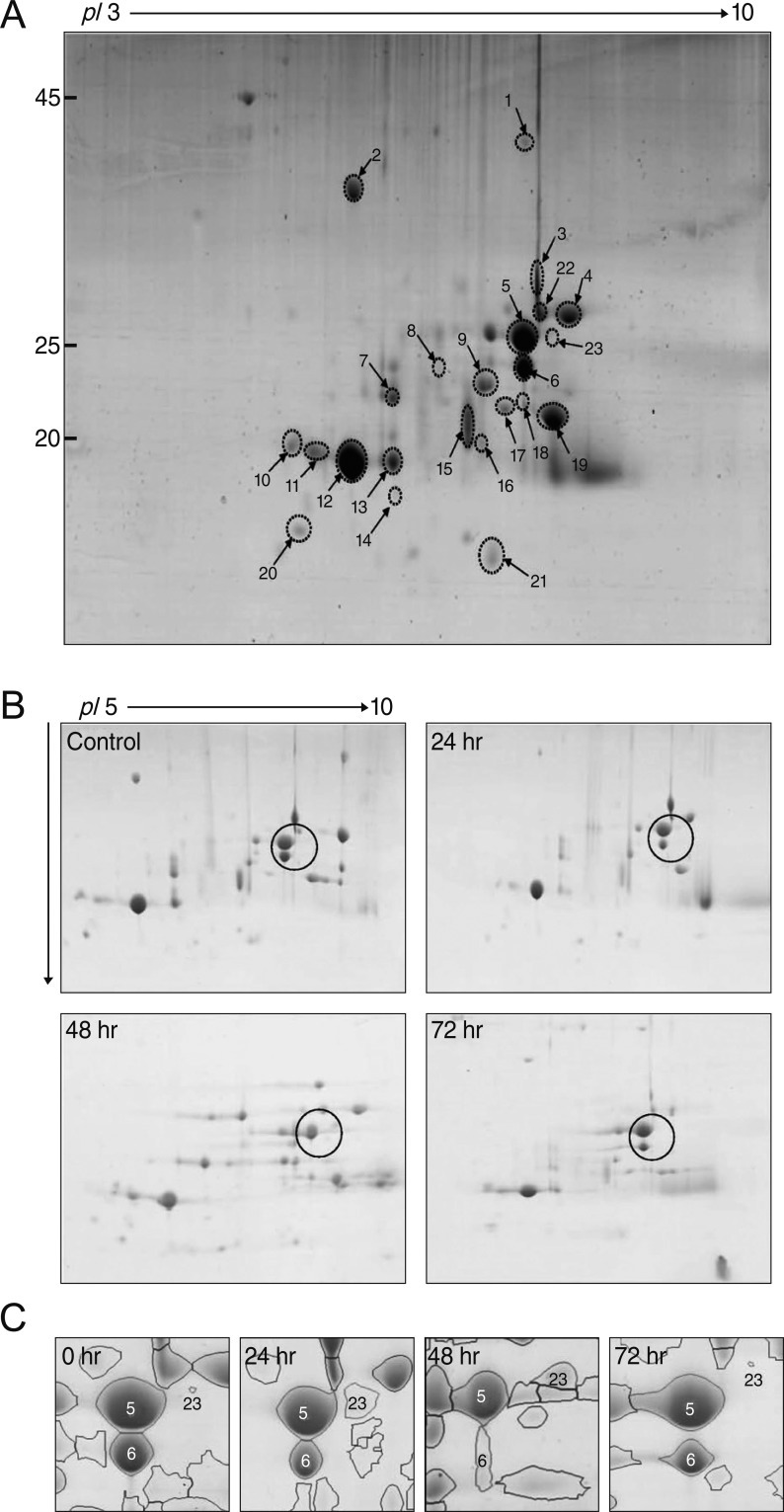
Identification of vitreous body proteins by two-dimensional electrophoresis gel MALDI-TOF/MS analysis
The vitreous bodies were subjected to 2-DE (Fig. 2), and the spots were analyzed using a MALDI-TOF/MS. Twenty-three spots were identified as crystallins (Table 1). As shown in the 2-DE gel images, spots 2, 10, 12, and 14 were αA-crystallin, while spots 17 and 19 were αB-crystallin. Spots 15, 16, and 18 were βA3-crystallin, while spots 7, 13, and 20 were βA4-crystallin. Spots 1, 5, 6, 9, and 23 were βB2-crystallin, while spots 4, 11, 21, and 22 were βB3-crystallin.
The 2-DE gel images of the control and at 24 hours, 48 hours, 72 hours after I/R injury are shown in Fig. 2, and the overall patterns are similar. The changes in all the spots are depicted in Fig. 3. The αA-crystallin and αB-crystallin had not changed at 24 hours; however, αA-crystallin and β-crystallin increased to the control level at 48 hours after I/R injury and declined again at 72 hours (Fig. 3A). Spot 15 of βA3-crystallin was significantly decreased at 24 hours but rapidly recovered to half of the control value. Spot 13 of βA4-crystallin was sustained at 24 hours but decreased dramatically at 48 hours and 72 hours after I/R injury. Spot 4 of βB3-crystallin was decreased at 24 hours and recovered to the control level at 72 hours after injury.
In terms of βB2-crystallin, spot 6 disappeared from the vitreous body within 48 hours after I/R injury. However, spot 23 was detected in the entire I/R injury model but not in the control sample. The truncated form (spot 6) of βB2-crystallin was critical in the normal control group. On the other hand, the intact form of βB2-crystallin (spot 23) was expressed in the I/R injured animals after 48 h (Fig. 2C).
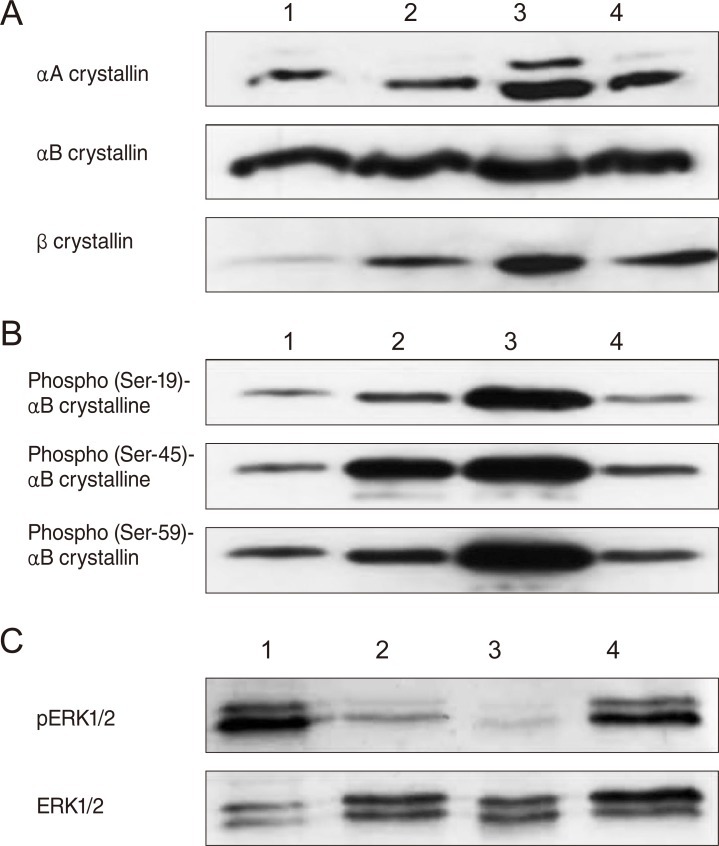
Western blot of vitreous bodies using anti-αA crystallin, anti-αB crystallin, and anti-β crystallin
We performed Western blotting of the extracted vitreous body proteins using anti-αA crystallin, anti-αB crystalline, and anti β-crystallin antibodies (Fig. 3A). The total αA crystallin increased at 48 hours and decreased at 72 hours after I/R injury. However, the total αB-crystallin showed no significant changes. The total β-crystallin showed dramatic changes compared to the αA and αB crystallins, increasing at 24 hours, peaking at 48 hours, and decreasing at 72 hours after injury.
Phosphorylation of αB crystallins
Phosphorylation of αB crystallins was observed at three serine sites (Ser-19, Ser-45, and Ser-59) by Western blotting (Fig. 3B). The phosphorylation at the Ser-19 and the Ser-59 of the αB crystallin was significantly increased at 48 hours after I/R injury and returned to the control level at 72 hours after injury. However, the phosphorylated Ser-45 of αB crystallin increased significantly at 24 hours and peaked at 48 hours after I/R injury.
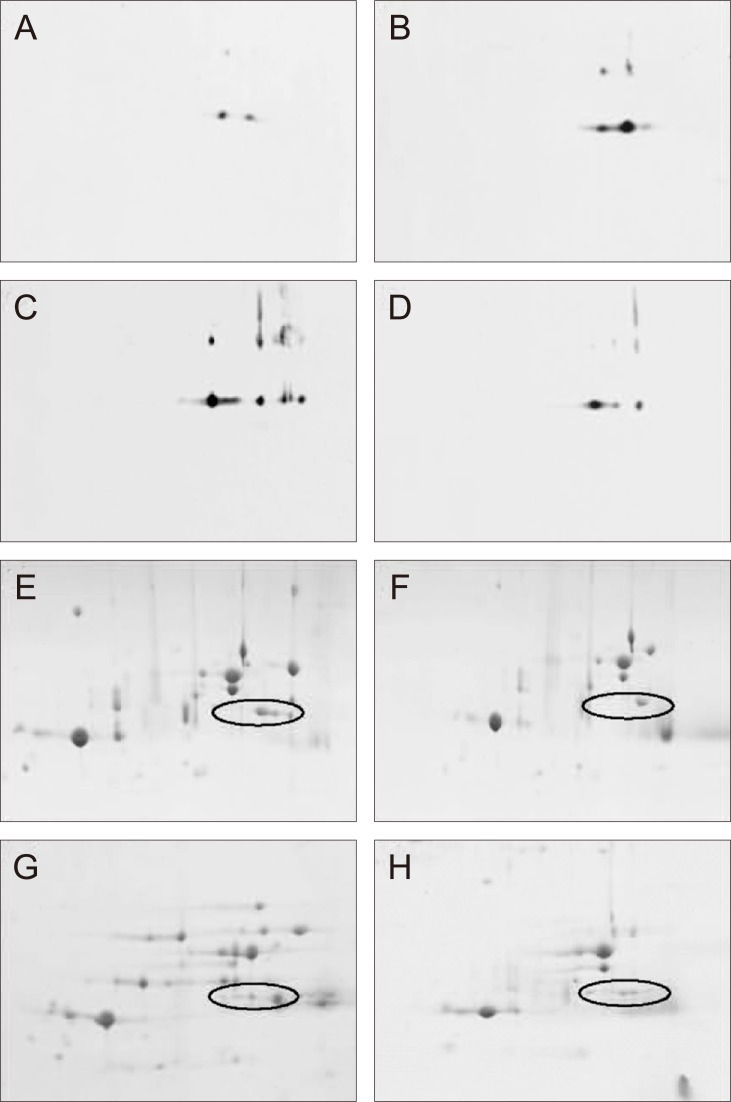
Western blotting of phospho-serine 45 of αB crystallin after two-dimensional electrophoresis
After the 2-DE, we performed Western blotting of phospho-serine 45 of αB crystallin (Fig. 4). The blotted area corresponded to spots 17 and 19 of the 2-DE gels (Fig. 2), and three distinctive spots were detected. Phosphorylation of Ser-45 of αB crystallin occurred at both spots 17 and 19, showing an increase at 24 hours, a peak at 48 hours, and then a decline at 72 hours after injury. The results were compatible with the Western blotting results of the phosphorylated Ser-45 of αB crystallin, as shown in Fig. 3B.
ERK1/2 phosphorylation
Western blotting was performed for ERK1/2 and phosphorylated ERK1/2 (pERK1/2) because the phosphorylated Ser-45 of αB crystallin had increased significantly 24 hours after I/R injury and peaked at 48 hours. The phosphorylated ERK1/2 was decreased after 24 hours and 48 hours and then recovered at 72 hours (Fig. 3C).
Discussion
I/R injury, the most common pathogenic mechanism in eye disease due to ocular inflammation, frequently damages the blood-retinal barrier [2]. In the current study, we used a proteomics methodology to investigate the crystallins of vitreous bodies obtained after I/R injury. The damage to the ganglion cell layer of the retina (Fig. 1) was most severe at 48 hours after I/R injury. However, there was a subsequent recovery at 72 hours. The responses of αB- and βB2-crystallins to I/R injury in the vitreous bodies of SD rats appear to be similar to those observed in inflammation and host defense. We identified the crystalline proteins by peptide mapping using MALDI-TOF/MS. The βB2-crystallin spots 5, 6, and 23 showed the most obvious changes (Fig. 2C).
The crystallins were initially thought to be lens proteins but have subsequently been revealed to be proteins that maintain the intracellular stability via chaperone activity and cell signaling pathways [22-24]. Unlike αB crystallin, αA and β-crystallins in the vitreous body increased at 48 h after I/R injury (Fig. 3A). In contrast, the phosphorylation of ERK1/2 declined up to 48 h and then recovered. The preferential phosphorylation of αB-crystallin at Ser-19, Ser-45, and Ser-59 (Fig. 3B) led to accelerated ERK1/2 inactivation at 24 and 48 hours after I/R injury (Fig. 3C). As a type of heat shock protein, αB-crystallin is ubiquitously expressed in several human diseases and is known to mediate a pro- or anti-inflammatory reaction [25-28]. In our previous studies, crystallins (the major proteins of the vitreous body) were induced in response to ocular inflammations of animal uveitis models. These findings suggested that post-translational truncations of αA- and αB-crystallins, phosphorylation of αB-crystallin, and new production of βA4- and βB2-crystallins are intercorrelated with uveitis progression and inflammatory responses [13,14]. The regulatory mechanism of the phosphorylation of αB-crystallin [29] results in the inhibition of RAS [23]. This finding indicates that the downstream inactivation of ERK1/2 is triggered by phosphorylated αB-crystallin. Therefore, phosphorylated αB-crystallin is expected to increase the RAS, which indicates that organs are protected from I/R injury, so it can be more obvious after the transplantation [30,31].
Taken together, our results suggest that phosphorylated αB crystallin may inhibit the RAS that causes inactivation of ERK1/2 (a downstream target of the raf-1 and MEK1/2 pathways). This inactivation is associated with suppression of the inflammation triggered by I/R injury (Fig. 5).

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print