The Na+-K+-2Cl--cotransporter (NKCC) belongs to the cation-chloride co-transporter family, which transports Na+, K+, and Cl- ions across the plasma membrane of cells with a stoichiometry of 1Na+ : 1K+ : 2Cl-. The NKCC plays an important role in salt secretion and absorption, cell volume regulation, and maintenance of intracellular Cl- concentrations [1-3]. Recently, the NKCC has been reported to be associated with the cell cycle, cellular proliferation, and apoptotic cell death in ischemic tissues [4-7].
Two isoforms of the NKCC have been identified. The NKCC1 isoform was initially discovered on the basolateral membrane of the shark rectal gland and is expressed in a wide variety of tissues, including epithelial cells and non-epithelial cells. The NKCC2 isoform is expressed mainly in the apical membrane of epithelial cells of the thick ascending limb of Henle's loop in the kidney [1-3].
It has been reported that the NKCC is related with cell damage in many ischemic models [8-11]. In cerebral ischemia, NKCC1 contributes to astrocyte swelling, causing excitatory amino acid release, resulting in neuronal cell death [8,9]. Thus, pharmacological inhibition of NKCC1 is neuroprotective in ischemic or traumatic injuries of the brain. During heart ischemia, sodium accumulates outside cells due to impairment of ion channels or transporters like Na+-K+-ATPase, Na+-H+ exchanger, and Na+-Ca2+ exchanger and enters into cells through the NKCC [10]. Inhibition of the NKCC can attenuate the ischemic rise in intracellular sodium and maintain higher levels of high energy phosphates, significantly improving functional recovery.
Previous studies of the retina demonstrated that the NKCC is not expressed in retinal ganglion cells except during developing development [12,13]. However, the presence of the NKCC2 and its role in the retina have not yet been described. To address these issues, we analyzed the expression of the NKCC2 in the retina and observed expression change after retinal ischemia induced by intraocular pressure elevation.
Materials and Methods
Induction of transient retinal ischemia
Thirty-five adult male albino Sprague-Dawley rats weighing 200 to 250 g were used for this study. Animals were anesthetized with 4% chloral hydrate (1 mL/100 g body weight). The pupils were dilated with 1% tropicamide eye drops. The intraocular pressure (IOP) was raised to 90 to 120 mmHg by cannulation of the anterior chamber with a 30-gauge needle connected to a hydrostatic pressure device, and the elevated IOP was maintained for 60 minutes. The applied pressure of 90 to 120 mmHg is in the range, or slightly above, systemic systolic pressure in the rat. Following removal of the cannula, recirculation began immediately and the IOP decreased to normal values within 5 minutes. Animals were sacrificed by an overdose of chloral hydrate at 12 hours, 24 hours, 3 days, 7 days, and 14 days after reperfusion. The anterior segments of the eyeballs were removed. For western blot analysis, retinal tissues were quickly dissected on an ice-cold plate, frozen on dry ice, and stored at -70℃. For immunocytochemistry and quantitative analysis, the eyecups were fixed by immersion in fixative (4% paraformaldehyde/0.2% picric acid in 0.1 M phosphate buffer [PB, pH 7.4]), for 2 to 3 hours. Following fixation, the retina was carefully dissected and transferred to 30% sucrose in PB, for 24 hours at 4℃. They were then frozen in liquid nitrogen, thawed, and rinsed in 0.01 M phosphate buffered saline (PBS, pH 7.4). The central region near the optic disc was excised, dehydrated in a series of graded alcohol, and embedded in wax.
Immunoblot analysis
Western blot analysis was performed on the retinal extracts, which were homogenized in 10 volumes of 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton ×-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 0.02% sodium azide, 1 mM PMSF, and 5 µg/mL leupeptin. Protein concentration for each sample was assayed by the Lowry method, in duplicate, and the results were averaged. Duplicate sets of protein standards containing 0, 1, 3, 5, 10, 20, 40, and 60 mg of bovine serum albumin were assayed by the same method. The results were averaged and graphed to produce a linear equation that was used to estimate the protein contents in the retinal extracts. The optical density of each sample was measured at 660 nm, using a spectrometer (Spectronic 20; Bausch & Lomb, Rochester, NY, USA). Tissue samples corresponding to 25 g of total protein were heated at 100℃ for 10 minutes with an equivalent volume of sample buffer (containing 4% SDS and 10% mercaptoethanol) and loaded onto 10% polyacrylamide gels. The proteins were electrotransferred to a nitrocellulose membrane in Tris-glycine-methanol buffer. The membrane was blocked for 1 hour at room temperature in a blocking solution containing 5% nonfat dry milk, 0.05% Tween-20, and PBS. The membrane was then incubated for 15 hour at 4℃ with a goat polyclonal antibody directed against NKCC2 (1 : 1,000; Chemicon, Temencula, CA, USA) in the blocking solution. The membrane was rinsed with 0.05% Tween-20 in PBS for three washes of 10 minutes and incubated for 1 hour at room temperature in a 1 : 200 dilution of biotinylated donkey anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA). The blot was washed three times for 10 minutes each and then processed for analysis using an enhanced chemiluminescence detection kit (Amersham, Arlington Heights, IL, USA).
Immunocytochemistry
For the immunocytochemistry of the NKCC2, 50-µm-thick vibratome sections, prepared as described above, were used. Endogenous peroxidase activity was blocked by placing the sections in 1.4% H2O2 in methanol for 10 minutes. The sections were then incubated in 10% normal serum (Vector Laboratories) in PBS for 1 hour at room temperature in order to block nonspecific binding sites. Sections were then incubated in a solution of antibodies to the NKCC2 (1 : 1,000) for 1 day at 4℃. After washing in PBS for 45 minutes (3 × 15 minutes), the retinal tissues were incubated for 12 hours in biotinylated donkey anti-rabbit IgG (1 : 50, Vector Laboratories) with 0.5% Triton X-100 at 4℃, rinsed in PBS, and subsequently incubated in Avidin-biotin-peroxidase complex (Vector Laboratories) in PBS for 1 day at 4℃. Retinas were rinsed in two changes of PBS and three changes of 0.05M Tris-HCl buffer (TB, pH 7.4) for 5 minutes at room temperature, and were then incubated in 0.05% 3,3-diaminobenzidine tetrahydrochloride in TB for 10 minutes. Hydrogen peroxide was added to the incubation medium to a final concentration of 0.01% H2O2, and the container was gently shaken as the reaction proceeded. After 1.2 minutes of reaction, as determined by the degree of staining, the reaction was stopped with several washes of TB and PB. The stained tissues were mounted onto gelatin-coated slides and coverslipped with glycerol.
Confocal laser scanning microscopy
To identify whether cells demonstrating NKCC2 immunoreactivity were ganglion cells, double labeling techniques using antibodies against the NKCC2 and Thy1.1, a specific marker for ganglion cells, were performed. Sections were incubated overnight in monoclonal Thy1.1 antibody (1 : 500; Chemicon, Temencula, CA, USA) with 0.5% Triton X-100 in 0.1M PB at 4℃. Sections were rinsed for 30 minutes with 0.01 M PBS, and the retinal tissues were incubated for 2 hours in biotinylated donkey anti-mouse IgG (1 : 50, Vector Laboratories), rinsed in PBS, and subsequently incubated for 2 hours in biotinylated donkey anti-mouse FITC (1 : 50, Vector Laboratories). After rinsing for 30 minutes with 0.01 M PB and for 30 minutes with 0.01 M PBS and with subsequent incubation for 1 hour in normal donkey serum, the retina was incubated overnight in anti-NKCC2 at 4℃. Sections were then rinsed for 30 minutes with 0.01 M PBS and incubated for 2 hour in biotinylated donkey anti-rabbit IgG (1 : 50, Vector Laboratories), rinsed in PBS, and incubated for 2 hour in biotinylated Cy-3 (1 : 500, Vector Laboratories).
To identify whether cells demonstrating NKCC2 immunoreactivity were horizontal cells, double labeling techniques using antibodies against the NKCC2 and calbindin, a specific marker for horizontal cells, were performed. After incubation overnight in monoclonal calbindin antibody (1 : 3,000, Chemicon) with 0.5% Triton X-100 in 0.1 M PB at 4℃, sections were incubated for 2 hours in biotinylated donkey anti-mouse FITC (1 : 50, Vector Laboratories), rinsed for 30 minutes with 0.01 M PB and 30 minutes with 0.01 M PBS, and then incubated for 2 hours in biotinylated donkey anti-mouse IgG (1 : 50, Vector Laboratories) again. We incubated retinal tissues overnight in biotinylated donkey anti-mouse FITC (1 : 50, Vector Laboratories) and rinsed for 30 minutes with 0.01 M PB and 30 minutes with 0.01 M PBS. In order to inhibit nonspecific reactions, sections were incubated in normal donkey serum for 1 hour. After overnight incubation in anti-NKCC2 at 4℃, sections were rinsed for 30 minutes with 0.01 M PBS. The retinal tissues were incubated for 2 hours in biotinylated donkey anti-rabbit IgG (1 : 50, Vector Laboratories) and then for 2 hours in biotinylated Cy-3 (1 : 500, Vector Laboratories).
Sections were analyzed using a Bio-Rad Radiance Plus confocal scanning microscope (Bio-Rad, Hemel Hempstead, UK), installed on a Nikon Eclipse E600 fluorescence microscope (Nikon, Tokyo, Japan). FITC and Cy3 signals were always detected separately. The FITC labeling was excited using the 488 nm line of the Argon ion laser and detected after passing an HQ513/30 (Bio-Rad) emission filter. For detection of the Cy3 signal, the 543 nm line of the green HeNe laser was used in combination with the 605 / 32 (Bio-Rad) emission filter. Images were imported into Adobe Photoshop ver. 7.0 (Adobe Systems, San Jose, CA, USA) and printed on photo paper (Seiko Epson, Tokyo, Japan). For presentation, all manipulations (brightness and contrast only) were carried out equally for all images.
Whole-mount immunostaining and semiquantitative analysis
For whole-mount immunostaining, the same immunocytochemical procedures described above were used, but with a longer incubation time. For semi-quantitative analysis of post-ischemic changes in the NKCC2 immunoreactive ganglion cells, the density (per unit area, 0.25 mm × 0.2 mm) of the labeled ganglion cells was counted in the mid temporal region of the whole-mount retinas. The number of labeled cells was divided by the unit area to obtain mean densities of labeled cells per mm2. Data are given as mean ± standard deviation. Statistical significance was assessed using one-way ANOVA.
Results
Western blot analysis of the Na+-K+-2Cl--cotransporter 2 levels
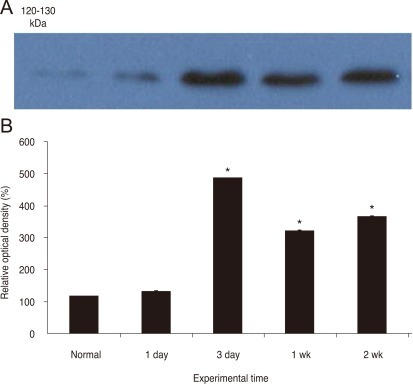
To evaluate the postlesional changes in the NKCC2 levels quantitatively, we performed an immunoblot analysis on membranes prepared from control and ischemic rat retina. As shown in Fig. 1, antibodies to the NKCC2 demonstrated a single band with a molecular mass of 100 to 150 kDa, the intensity of which increased up to 3 days following ischemia/reperfusion and gradually declined thereafter, in accordance with the immunocytochemical observations. Densitometric analysis revealed that the NKCC2 level was upregulated to approximately 115% of controls by 1 day and to approximately 415% by 3 days. The NKCC2 levels decreased to 275% and 313% at 7 and 14 days post ischemia/reperfusion, respectively.
The Na+-K+-2Cl--cotransporter 2 is expressed in the retinal ganglion cell layer and the outer plexiform layer
The thickness of the inner retina gradually decreased with increasing time after ischemia, in agreement with previous reports [14-16]. In normal rat retina immunostained for the NKCC2, labeling was mainly observed in the ganglion cell layer and outer plexiform layer (Fig. 2). In the ganglion cell layer, axonal staining was stronger than cell body staining. In the outer plexiform layer, labeled profiles were horizontally oriented. After ischemia/reperfusion, the NKCC2 immunoreactivity increased compared with controls.
The Na+-K+-2Cl--cotransporter 2 is expressed in retinal ganglion cells and horizontal cell processes
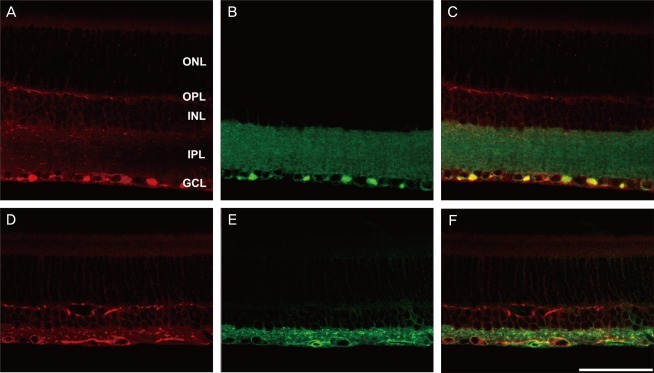
To identify the nature of the labeled profiles in the ganglion cell and outer plexiform layers, double labeling experiments using antisera against the NKCC2 were performed with anti-Thy1.1 for the ganglion cell layer and anti-calbindin for the outer plexiform layer. In the merged picture (Fig. 3C and 3F), the NKCC2 labeled profiles in the ganglion cell layer also demonstrated Thy1.1 immunoreactivity. Two weeks after injury, the ischemic retina (Fig. 3D-3F) demonstrated a decreased ganglion cell population compared with the normal retina (Fig. 3A-3C). In the other merged picture (Fig. 4C and 4F), the NKCC2 labeled profiles in the inner retina also demonstrated calbindin immunoreactivity. Two weeks after injury, the ischemic retina (Fig. 4D-4F) demonstrated increased horizontal cell process immunoreactivity compared with the normal retina (Fig. 4A-4C).
In the present study, all labeled profiles that lay in the ganglion cell and outer plexiform layers of the control retina and retinae of experimental groups were immunoreactive to antibodies against Thy1.1 and calbindin, respectively. These findings clearly indicate that the NKCC2 is expressed in retinal ganglion and horizontal cells.
The Na+-K+-2Cl--cotransporter 2 labeled ganglion cell population changes after ischemia/reperfusion
The density of the NKCC2-labeled ganglion cells was calculated in unit areas (0.25 mm × 0.2 mm) in the mid-temporal region of the retina (Figs. 5 and 6). In the normal retina, the mean density of labeled ganglion cells was 1255 ± 109 cells per unit area. These values correspond to the previous report of Lafuente et al. [14], Lafuente et al. [15], and Lafuente et al. [16]. The density of labeled cells was altered at 3 days and 2 weeks after reperfusion. The density of the labeled cells was 391 ± 49 (31.2% of normal density) and 185 ± 37 cells/unit area (14.7%) by 3 days and 2 weeks, respectively. These results are in agreement with a prior report by Lafuente et al. [14], Lafuente et al. [15], and Lafuente et al. [16], which demonstrated that retinal ganglion cell loss after retinal ischemia is an ongoing process and that most ganglion cells die after ischemia/reperfusion.
Discussion
Retinal ganglion cells express a variety of enzymes, neurotransmitter receptors, and voltage-gated ion channels. In circumstances in which these cells are severely stressed, cell injury results. Ca2+ ions are important mediators of cell injury and serve as an ubiquitous intracellular second messenger. Cytosolic Ca2+ ion concentration is maintained at a low concentration compared with extracellular Ca2+ ions. The Ca2+ concentration gradient determines the activity of many enzymes, through activation or inactivation of enzymes such as phospholipases, ATPases, proteases, and endonucleases. Increased intracellular Ca2+ levels induce apoptotic cell death [17-19].
During ischemia, total neuronal cell calcium content may increase to 150% or more compared to controls. Intracellular Ca2+ overloads and activates multiple molecular cascades, which contribute to eventual cell death. Although cell injury results from increased Ca2+ ion concentration, and this in turn mediates irreversible cell death cascades, loss of calcium homeostasis is not always an upstream event. The Ca2+ overload leading to cell death is normally preceded by elevation of intracellular sodium levels, caused by inhibition of the Na+/K+-pump, activation of the Na+/H+ exchange, and Na+-HCO3--cotransport following ATP depletion and acidosis. Reversal of the Na+/Ca2+ exchanger also contributes to intracellular Ca2+ increase. A significant component of axon loss after anoxic injury is due to the reverse operation of the Na+/Ca2+ exchanger triggered by the Na+ influx. The NKCC also contributes to excessive Na+ influx, and persistent Na+ influx accelerates neuronal damage by favoring Ca2+ influx [1-10]. So, theoretically Na+ channel blocking is more effective than calcium channel blocking for preventing these processes [20].
The NKCC is believed to play a critical role in the regulation of extracellular fluid volume and osmolarity. The NKCC1 isoform has been found in a variety of tissues, but the NKCC2 isoform has been identified only in the medullary regions of the kidney. Mutations of the NKCC2 have been reported to be linked to Batter's syndrome, a severe human genetic disease involving hypokalemic alkalosis with hypercalciuria.
In the brain, the NKCC1 maintains sodium concentration under physiological conditions. However, under pathophysiological conditions, such as ischemia, the NKCC1 contributes to an excessive sodium influx, which may induce cell damage via several mechanisms [8]. Mechanisms of acute ischemic injury in developing white matter astrocytes are not associated with Ca2+ influx, but are associated with the NKCC [9]. In ischemic myocardium in diabetics, alteration of the myocardial NKCC permeability results in increased entry of Na+. Inhibition of the NKCC with butmetanide reduced ischemia/reperfusion injury by 50% in diabetic hearts [10]. Such examples suggest key roles of the NKCC in ischemic cellular injury.
In the case of retinal ganglion cells, there have been no reports regarding the existence of the NKCC. As such, there has been no research on the role of the NKCC in retinal ischemia. Previous studies demonstrated that the NKCC is expressed in horizontal cells, starburst amacrine cells, and on bipolar dendritic tips, but not in ganglion cells [12,13]. Only in the developing retina is the NKCC transiently expressed on the ganglion cells. However, these previous studies were performed only in regards to NKCC1; there have been no prior reports on NKCC2 in the retina.
In the normal rat retina, light photomicrographs of vibratome sections demonstrated that NKCC2 labeling cells were observed mainly in the ganglion cell and outer plexiform layers. Axon bundles of ganglion cells are more immunoreactive than cell bodies. Confocal laser scanning microscopy with Thy1.1 and calbindin revealed ganglion cells and horizontal cells as the sources of the NKCC2, respectively. Ganglion and horizontal cell processes have strong NKCC2 immunoreactivity. The NKCC2 was identified as a good marker of retinal ganglion cells, similar to Thy1.1, which has been widely used. Additionally, retinal damage models demonstrated that both protein and mRNA levels of Thy1.1 change to a much greater extent than RGC number loss [21]. As the NKCC2 immunoreactivity increases during ischemia, it can be used as a marker of retinal ganglion cell stress.
Following ischemia/reperfusion, immunoblots of the NKCC2 protein levels increased and reached a peak of approximately 415% of control levels 3 days after ischemia/reperfusion. There is a direct relationship between the duration of insult and the quantity of neuron loss [14]. We raised intraocular pressure to 120 mmHg for 60 minutes. According to prior studies that used a similar experimental paradigm, cellular reaction to ischemic insult has a peak at 3 days [22,23].
As reported in prior research on the quantification of retinal ganglion cell death after 120 minutes of ischemia, 95% of ganglion cells were lost between 5 and 30 days after the insult [24]. The current study demonstrated that the mean density of NKCC2 labeled ganglion cells per unit area (0.25 mm × 0.2 mm) decreased from 1,255 ± 109 to 391 ± 49 (31.2% of normal density) by 3 days and 185 ± 37 (14.7% of normal density) by 2 weeks.
In the current study, although both the ganglion and horizontal cell processes expressed the NKCC2, only the number of ganglion cell decreased; the number of horizontal cells did not decrease. There are several causes for selective cell death of ganglion cells compared with horizontal cells.
N-methyl-D-aspartate (NMDA) toxicity to retinal ganglion cells is well known. As the retinal ganglion cell express NMDA receptors, they are vulnerable to ischemic injury [25]. Inner retinal neurons are also susceptible to ischemic insults as both ionotropic and metabotropic receptors are widely expressed in inner retinal neurons [26]. Because the glutamate receptors expressed on horizontal cells are AMPA receptors [27], they may not have a major role in ischemic cell death. Calcium binding protein is also important; these cells are resistant to ischemia through calcium buffering. Calbindin-labeled horizontal cells are resistant to ischemic insults in the rat retina [28]. Calbindin, an EF-hand calcium binding protein, controls the level of intracellular calcium within the first 100 milliseconds following calcium influx in retinal horizontal cells [29]. Therefore, rat horizontal cells would more resistant to injury due to their greater capacity to buffer intracellular calcium induced by excitotoxicity. Moreover, excessive Ca2+ influx triggers formation of nitric oxide (NO) via neuronal NO synthase (nNOS). In ischemic retina, Muller and retinal ganglion cells express nNOS and inducible NOS. Also, matrix metalloproteinase is activated via neuronal nitric oxide synthase [30,31].
There are few prior reports on horizontal cell changes in ischemia/reperfusion. In the current study, horizontal cell specific immunostaining demonstrated marked axonal sprouting from early phases. Retinal horizontal cells are a famous and long-standing example of gap junctional coupling in all species [32,33]. We assumed that marked axonal sprouting may be due to the gap junction and horizontal cell axonal sprouting may explain the elevated NKCC2 expression. We attempted to investigate the increase of the NKCC2 expression in horizontal cells, but this was not technically feasible as the antibody to the NKCC2 did not penetrate well into the outer plexiform layer.
In summary, we have presented three major findings. First, the NKCC2 is expressed in retinal ganglion and horizontal cells processes. Second, the NKCC2 expression was upregulated in the ischemic rat retina; the NKCC2 immunoreactivity increased after ischemia/reperfusion compared with controls. Third, the number of NKCC2-labeled ganglion cells decreased after retinal ischemia. Our findings lead us to propose that treatment of ischemic insults, such as glaucoma, with inhibitors of the NKCC2 warrants further study.




 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print