Comparison of Clinical Manifestations between Patients with Ocular Myasthenia Gravis and Generalized Myasthenia Gravis
Article information
Abstract
Purpose
To compare the clinical manifestations between patients with ocular myasthenia gravis and those with generalized myasthenia gravis (MG).
Methods
The medical records of 71 patients diagnosed with MG between January 1995 and December 2007 were reviewed. Demographics, sensitivities of diagnostic methods, the presence of systemic autoimmune diseases, ophthalmic complications caused by MG, and treatments were evaluated and compared.
Results
Fourteen patients (20%) were diagnosed with ocular MG and 57 patients (80%) with generalized MG. Sensitivities of anti-acetylcholine receptor antibody and repetitive nerve stimulation tests were significantly higher in the generalized MG group (84%, 89%) compared to those in the ocular MG group (50%, 54%) (p = 0.011, p = 0.008). The sensitivity of the neostigmine test was the highest in both groups (98% of generalized MG, 79% of ocular MG), and the difference between the two groups was borderline significant (p = 0.058). The most common symptoms were ptosis and diplopia, and both groups presented with pain, blurred vision, and tearing. Systemic autoimmune disease was more prominent in the generalized MG group (21%) than in the ocular MG group (14%), and steroid therapy was used more frequently in the generalized MG group (82%) than in the ocular MG group (57%). Ophthalmic complications associated with long-term steroid treatment were more profound in the generalized MG (30%) compared to those of the ocular MG (21%).
Conclusions
The generalized MG group was associated with higher sensitivities to diagnostic tests, more systemic steroid use, higher ophthalmic complications caused by systemic autoimmune disease, and long-term steroid treatment compared to those of the ocular MG group.
Myasthenia gravis (MG) is an autoimmune disease characterized by muscular fatigue due to defective neuromuscular transmission, with the levator palpebralis and extraocular muscles preferentially affected [1]. More than three-quarters of MG patients present with visual complaints such as droopy eyelids or double vision, and about half of patients who present with ocular manifestations develop generalized weakness within six months. Approximately 80% of MG patients will generalize within two years and approximately 90% within three years [2-5]. The first and sometimes only manifestations of MG are ocular including fluctuating diplopia and/or ptosis worsened by exertion. Several retrospective studies have suggested that early treatment with oral prednisolone can delay the onset and possibly decrease the frequency of progression from ocular MG to generalized MG. High-dose oral prednisolone over a period of two to three months is generally required to induce remission [6-12].
MG patients are more likely to develop other immune-related disorders including type I diabetes mellitus, Graves disease, Hashimoto disease, systemic lupus erythematosus, rheumatoid arthritis, Sjogren syndrome or multiple sclerosis [11,12]. These autoimmune diseases may be associated with ophthalmic symptoms including blurry vision, tearing, pain, photophobia, foreign body sensation, and ophthalmic signs including reduced visual acuity, decreased tear film, inflammatory soft tissue, exophthalmos, and lid retraction that can occur along with the typical MG symptoms of ptosis and ocular motility defects.
To date, previous studies of MG have not focused on ophthalmic manifestations other than ptosis, diplopia and those caused by the long-term treatment of MG. There are no known studies comparing the ophthalmic features and complications between patients with ocular MG and those of generalized MG. Therefore, we compared the demographics, clinical ophthalmic features of MG, sensitivities of MG diagnostic methods, systemic treatment of MG, the presence of systemic autoimmune diseases, and the ophthalmic complications caused by MG itself, other associated autoimmune diseases and long-term steroid treatment between ocular and generalized MG patients.
Materials and Methods
Patients diagnosed with MG between January 1995 and December 2007 with a minimum follow-up period of one year were selected from the database of the Department of Neurology and Ophthalmology, Yonsei University College of Medicine. Data from the early phase of the disease and diagnosis were collected from medical charts with an emphasis on demographic data, ophthalmic symptoms and signs, other associated autoimmune diseases, and the results of diagnostic tests. The diagnosis of MG was determined primarily through clinical evaluation and confirmatory diagnostic tests including one or more of the following: anti-acetylcholine receptor antibody test, repetitive nerve stimulation test, neostigmine test and significant response to a longer acting acetylcholinesterase antagonist (pyridostigmine). Sensitivities of the anti-acetylcholine antibody test, repetitive nerve stimulation test, and neostigmine test were evaluated and compared between ocular MG and generalized MG groups.
Patients were classified according to the modified Osserman score: grade I, focal disease (ocular MG, restricted to the ocular muscle); grade II, generalized disease that is either mild (IIa) or moderate (IIb); grade III, acute severe generalized disease with respiratory failure; and grade IV, severe generalized disease with respiratory failure (progression within two years) [13]. Patients were classified into two groups of ocular (grade I) and generalized MG (grade II-IV). All patients underwent contrast-enhanced computed tomography of the chest to screen for thymoma. Patients with radiologically-diagnosed thymoma were operated on if their general condition allowed for surgical intervention. The ophthalmic symptoms and signs associated with MG including ptosis, diplopia, and caused by long-term steroid treatment were compared between the ocular MG and generalized MG groups. Statistical analysis was performed using SPSS ver. 12.0 (SPSS Inc., Chicago, IL, USA). Categorical variables were analyzed using the Pearson's chi-square and the Fisher's exact tests, while continuous variables were analyzed with the Wilcoxon rank sum test. A p-value of less than 0.05 was considered statistically significant.
Results
A total of 71 patients with a median of 39 months (range, 12 to 105 months) follow-up were included in this study. The mean age was 48.5 years (standard deviation [SD], 16.1 years; range, 7 to 80 years), and the female to male ratio was 1.56:1. The mean age at disease onset was 41.8 years (SD, 16.9 years; range, 3 to 71 years), and initial symptoms developed before the age of 50 in 65% (46/71) of MG patients.
Using the modified Osserman score, patients were classified as I (20%), IIa (16%), IIb (39%), III (21%), and IV (4%). Fourteen patients presented with ocular MG and 57 patients with generalized MG. In patients with generalized MG, the mean time interval from the onset of ocular MG to generalization was 3.5 years. Table 1 presents the comparative demographic and clinical characteristics. Age, gender, and age of disease onset were not significantly different between the ocular MG and generalized MG groups. Thymoma was radiologically diagnosed and surgically resected in 40 patients (56%, 27 women and 13 men), with 22 cases of histologically confirmed thymoma postoperatively (ocular MG, 3 patients; generalized MG, 19 patients). Thymectomy was performed significantly more in the generalized MG group (65%) than in the ocular MG group (21%) (p = 0.003, Fisher's exact test). The remaining 18 patients with generalized MG had hyperplastic thymus (n = 14) or normal histology (n = 4).
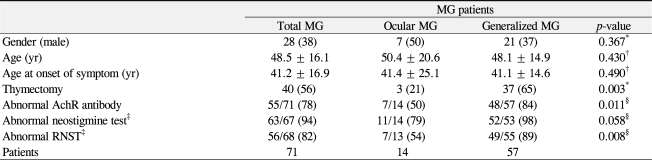
Demographic characteristics in myasthenia gravis (MG) patients
Fig. 1 presents the distribution for age of disease onset according to gender. The criteria in Fig. 1 are based on the report by Beekman et al. [14]. Mean age of disease onset was 40.1 years (SD, 13.6 years; range, 3 to 67 years) in male MG patients and 42.8 years (SD, 13.9 years; range, 3 to 69 years) in female MG patients. The ages of disease onset were not significantly different between female and male MG groups (p = 0.693, Wilcoxon rank sum test).
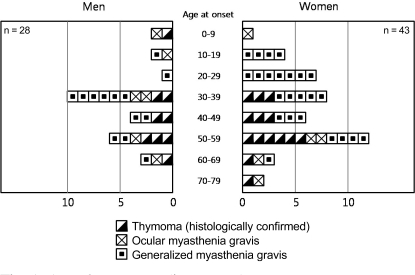
Age of onset according to gender.
The anti-acetylcholine receptor antibody test was performed in 71 patients, repetitive nerve stimulation test in 68 patients and neostigmine test in 67 patients (Table 1). The sensitivities of the anti-acetylcholine receptor antibody and repetitive nerve stimulation tests were significantly higher in the generalized MG group (84%, 89%, respectively) compared to those in the ocular MG group (50%, 54%, respectively) (p = 0.011, p = 0.008, respectively). The sensitivities of the neostigmine tests were higher in both groups (98% in the generalized MG group and 79% in the ocular MG group) compared to those of the other tests, and the difference between the two groups was borderline significant (p = 0.058).
The ophthalmic symptoms and signs in the ocular MG group and the generalized MG group are provided in detail in Table 2. In the total 71 patients, 87% presented with fluctuating ptosis and 93% presented with diplopia. Although 100% of the ocular MG group presented with ptosis or diplopia, 7% of the generalized MG group had no ptosis or diplopia. Interestingly, other ophthalmic symptoms including ocular pain, blurred vision, and tearing were observed in both MG groups (24% to 41%); however, the incidences were not significantly different between the ocular MG group and the generalized MG group. Ophthalmic signs such as dry eye, exposure keratitis, lid retraction, lid swelling, lid erythema, cataract, glaucoma, and central serous chorioretinopathy were present and were associated with muscle weakness caused by MG, systemic autoimmune diseases, and/or long-term steroid treatment. One patient from the generalized MG group presented with exposure keratitis due to poor eyelid closure caused by facial weakness. Nineteen patients (21% of ocular MG and 28% of generalized MG) were diagnosed with dry eye syndrome according to the Schirmer test (<10 mm) and tear break up time (less than 10 seconds) with dry eye symptoms such as pain or tearing. One patient with dry eye was diagnosed with Sjogren's syndrome with documented elevated levels of anti-nuclear antibodies including SSA/Ro and SSB/La. Patients with lid retraction (n = 2), lid swelling or erythema (n = 3) were diagnosed with thyroid-associated ophthalmopathy and presented with generalized MG, except for one patient who presented with ocular MG. Five patients (ocular MG, 1; generalized MG, 4) were diagnosed with open angle glaucoma after long-term steroid treatment for MG and presented with visual field defects and required anti-glaucoma eye drops or filtering surgery to control intraocular pressure. Twelve patients (ocular MG, 1; generalized MG, 11) presented with cataract after the diagnosis of MG, nine of whom had posterior subcapsular cataract and three of whom had mixed type of cataract. During the follow-up period, three of those 12 patients required cataract surgery. Three patients with a history of steroid treatment for a mean of 6.1 years developed central serous chorioretinopathy; one patient developed central serous chorioretinopathy immediately after intravenous steroid pulse therapy.
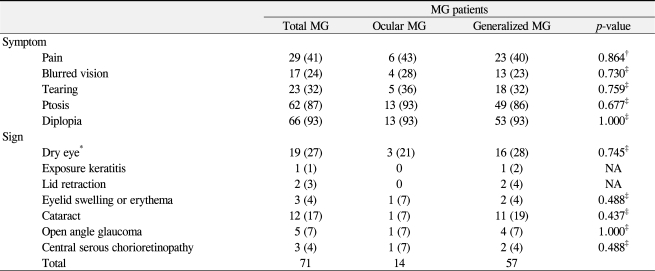
Ophthalmic symptoms and signs in myasthenia gravis (MG) patients
As presented in Table 3, associated autoimmune diseases were present in 14 patients (20%, seven women and seven men), and although the generalized MG group had a higher incidence (21%, 12/57) compared to that of the ocular MG group (14%, 2/14), the difference was not statistically significant (p = 0.721). Five patients had ocular involvement of Graves hyperthyroidism with abnormal thyroid stimulating antibody and thyroid hormone levels; one patient required three-wall decompression surgery for compressive optic neuropathy. Four patients (6%) had hypothyroidism without ophthalmopathy, diagnosed as Hashimoto's thyroiditis. The other associated diseases were rheumatoid arthritis (n = 1), systemic lupus erythematosus (n = 1), multiple sclerosis (n = 1), Sjogren's syndrome (n = 1), and Behcet's disease (n = 1). Among the 12 patients with autoimmune diseases, all except two patients with Graves' hyperthyroidism and Hashimoto's thyroiditis presented with generalized MG.

Associated autoimmune diseases
Sixteen patients were treated with only an anticholinesterase drug such as pyridostigmine or dosmin (Table 4). The percentage of patients who were only treated with acetylcholinesterase inhibitor was much higher in the ocular MG group (44%) than in the generalized MG group (20%), although the difference was not statistically significant (p = 0.07). Prednisolone was administered orally to 55 patients (77%) who were not controlled with pyridostigmine; this low-dose prednisone was maintained until complete remission. Twenty-three of the 55 patients received high dose intravenous steroid pulse therapy for myasthenic crises. Prednisolone and intravenous high dose steroid pulse therapy were administered to the generalized MG patients (82% and 35%) more frequently than it was to the ocular MG group (57% and 21%, respectively), although the differences were not statistically significant (p = 0.07, p = 0.525, respectively). Two generalized MG patients in myasthenic crisis required plasmapheresis. Two ocular MG patients (14%) and 22 generalized MG patients (39%) were treated with prednisolone combined with another non-steroidal immunosuppressant such as azathioprine, cyclosporine and cyclophosphamide.
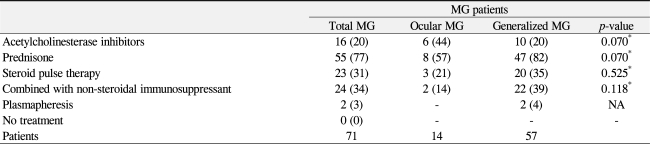
Treatment modalities of myasthenia gravis (MG)
Discussion
This is the first study comparing the ophthalmic manifestations and complications associated with MG, systemic autoimmune disease and long-term steroid treatment. As MG is a rare disease, it is often difficult to analyze a large patient population. We evaluated 71 patients that were followed for at least one year and analyzed and compared the demographics, clinical manifestations, diagnoses and treatment modalities between the ocular and generalized MG groups.
The most common symptoms seen in patients with MG are ptosis and diplopia. As expected, the majority of the ocular MG and generalized MG groups presented with ptosis (87%) or diplopia (93%). Eleven percent (8/71) of the generalized MG patients had only ptosis, 17% (12/71) experienced only diplopia, while the majority (76%, 54/71) suffered from both ptosis and diplopia. In the ocular MG group, less than 10% had ptosis only (1/14) or diplopia only (1/14), while 79% (12/14) had both ptosis and diplopia. A significant number of patients reported various ophthalmic symptoms such as tearing (32%), pain (41%), and blurred vision (24%), all of which may be related to the muscular weakness caused by MG. Interestingly, these symptoms were observed equally in the ocular and the generalized MG groups. Since patients with MG have difficulty blinking, dryness, irritation and weakness may occur. Weakness of the orbicularis muscles that control eye closure may result in excessive tearing due to incomplete blinking. Artificial tears and forced blinking may help to reduce these ocular-irritating symptoms.
The neostigmine test has a relatively high sensitivity, 94% to 100% for generalized MG and 69% to 91% for ocular MG [14]. Consistent with previous studies, the results of the neostigmine test presented sensitivity rates of 98% for generalized MG and 79% for ocular MG in this study. In addition, 78% of MG-diagnosed patients were positive for anti-acetylcholine receptor antibody (50% in ocular MG, 84% in generalized MG), in agree with the results of previous studies, which have reported sensitivities ranging from 73% to 93% in MG patients [14-16]. When the repetitive nerve stimulation test was performed in 68 patients, the positive rate was low in ocular MG patients (54%) and relatively high in generalized MG patients (89%). The repetitive nerve stimulation test has been shown to have a high sensitivity (74% to 84%) for generalized MG but a low sensitivity (22% to 36%) for ocular MG [17]. In conclusion, the basic diagnostic methods for MG had lower sensitivities in the ocular MG group compared to those in the generalized MG group, in agreement with previous reports. The ice test, in which sensitivities are reported to be 80% to 94% [18,19], may be a good alternative diagnostic method; however, we could not compare the results of the ice test between the ocular and generalized MG groups as this test was performed in only a small number of patients with ocular MG (data not presented).
A bimodal pattern of age of onset has been recognized in both genders of MG patients, with an early-onset peak between ages 20 to 30 years and a late-onset peak between ages 70 to 75 years [20]. Incidence rates in both genders increase with age, peaking between 60 to 80 years, with an apparent male predominance in the older age group [21]. In this study, the mean age of disease onset of was 40.1 years in male MG patients and 42.8 years in female MG patients, and the peak incidence was between ages 30 to 39 years in males and between 50 to 59 years in females, as presented in Fig. 1.
Twenty percent (n = 14) of the patients had autoimmune diseases associated with MG, with thyroid diseases being the most frequent (13.0%). Epidemiological studies have shown that thyrotoxic myopathy resulting from autoimmune thyroid disease occurs in approximately 5% to 10% of patients with MG [22], and similar findings were reported in a study from Denmark [23]. Five patients presented with symptoms of thyroid-associated ophthalmopathy (7.0%, one ocular MG, four generalized MG). Patients also presented with associated autoimmune diseases such as rheumatoid arthritis, Sjogren's syndrome, multiple sclerosis, and Behcet's disease. Although statistical comparison of incidences of autoimmune diseases between the ocular and generalized MG groups was not possible due to the small sample size, the incidence of total autoimmune disease was higher in patients in the generalized MG group (21%, 12/57) than it was in the ocular MG group (14%, 2/14). The prevalence of concurrent autoimmune diseases in a previous study was also higher in the generalized MG group (25%, 3/12) compared to that in the ocular MG group (19%, 5/27) [23].
Both ocular and generalized MG patients experience remissions and exacerbations on a regular basis. Treatment medications include anticholinesterase inhibitors, steroids and other immunosuppressants either alone or in combination. Thymectomy is often recommended for generalized MG patients; however, this procedure is rarely performed in purely ocular MG patients [24]. Steroid treatment is recommended as the first-line drug in all patients with generalized MG who do not respond to thymectomy [25]. Anticholinesterase drug monotherapy cannot induce remission of generalized MG and may mask ongoing autoimmune damage. Concurrent steroid treatment is therefore required in these cases [10]. Steroid therapy was used more frequently in generalized MG patients (82%) than in patients with ocular MG (57%), although the difference was not significant. In cases of myasthenic crisis, various treatments such as pulse steroid therapy, plasmapheresis, and intravenous immunoglobulin therapy can be considered [24]. High dose pulse steroid treatment was performed more frequently in the generalized MG group (35%) than it was in the ocular MG group (21%), although the difference was not statistically significant (p = 0.525). A previous study reported that nearly all ocular MG patients (93%) illustrated some symptomatic improvement after anticholinesterase monotherapy [7], although ptosis has been shown to respond better to to this treatment than does diplopia [8]. In another recent study conducted in Thailand, ocular MG patients presenting with initial symptoms of ptosis and diplopia showed a decreased response to anticholinesterase treatment compared to another MG group presenting without initial symptoms of ptosis and diplopia [26]. Anticholinesterase drugs improve symptoms of MG in nearly all ocular and generalized MG patients, but the therapeutic effects are limited. Thus, most patients require additional immunosuppressive treatment such as corticosteroid treatment [27]. Although still controversial, there are several studies which have insisted that systemic steroid treatment in ocular MG patients can prevent disease progression to generalized MG [6,8,9,11,12,28]. In this study, 57% of the ocular MG group received steroid treatment since anticholinesterase drug monotherapy alone did not induce complete remission.
Four of 71 patients presented with open angle glaucoma, and all of them were receiving long-term corticosteroid treatment to induce remission (mean, 4.7 years). Corticosteroids increase the risk of glaucoma by increasing the intraocular pressure when administered exogenously (topically, periocularly or systemically) and in certain conditions of increased endogenous production (e.g., Cushing's syndrome) [29]. In addition, prolonged use of corticosteroids is a significant risk factor for the development of posterior subcapsular cataract [30]. Nine of 12 patients who presented with cataract had posterior subcapsular cataract, and three of 12 cataract patients required cataract surgery.
Interestingly, three patients developed symptomatic central serous chorioretinopathy during steroid treatment for MG, one of the least known complications of steroid treatment. Although idiopathic central serous chorioretinopathy is known to be mild with spontaneous recovery and minimal long-term effects on visual acuity, irreversible visual impairment may occur as a result of long-term steroid therapy [31]. Although one of the three central serous chorioretinopathy patients recovered without treatment, the other two patients showed serous detachment of the neurosensory retina, confirmed by optical coherence tomography at the final follow-up visit, and the final visual acuities of the central serous chorioretinopathy-affected eyes were <20/100.
Systemic complications associated with steroid treatment include aseptic bone necrosis, hyperglycemia, osteoporosis, immune compromise, hypertension, growth retardation in children and proximal myopathy [32], with most complications correlating with the cumulative dose. Children and the elderly are particularly susceptible to corticosteroid complications. All three pediatric patients had short stature, and osteoporosis was diagnosed in six of the eight elderly patients (>60 years) during the follow-up period. An important method for preventing complications in patients receiving corticosteroids is to limit the total steroid dose. One way to reduce the steroid dose is by performing early thymectomy during the course of treatment. Thymectomy is accepted as an effective treatment for MG [33], for which it is considered a first line immune treatment [10]. Thymectomy was performed in 65% of generalized MG patients, and the majority was able to reduce steroid dosage while continuing symptom improvement.
To date, MG has been predominantly researched by neurologists, and as a result, ophthalmic complications other than ptosis and diplopia have not been thoroughly researched. Neurologists may not detect ocular symptoms and signs other than ptosis and diplopia or may not be aware of the ophthalmic complications that can result from combined autoimmune diseases and steroid treatment. As a result, MG patients with ophthalmic signs and symptoms may not receive immediate proper ophthalmic management. We evaluated ophthalmic complications in addition to ptosis and diplopia in relation to the presence of autoimmune diseases and long term steroid treatment. It is important that patients with ocular MG or generalized MG with ophthalmic symptoms should undergo regular eye examinations. In cases of MG, the neurologist should recommend an eye specialist who is well aware of the ophthalmic complications of the disease and its prognosis. Most importantly, careful attention must be given to generalized MG patients because they have a higher risk of ophthalmic and other problems associated with systemic autoimmune disease and long-term treatment of MG than do ocular MG patients.
Selection bias, which is commonly found in retrospective studies, was a potential problem in the current study as our analysis was limited to information available from medical records. It was impossible to quantitatively compare the treatment efficacies of anticholinesterase inhibitors and steroids or to objectively evaluate the severity of ptosis and diplopia due to fluctuations in disease status and treatment response.
In conclusion, MG patients may experience less common eye problems including dry eye symptoms, pain, blurry vision, and tearing in addition to more well-known symptoms such as ptosis and diplopia. These less common eye problems can impair quality of life and should not be neglected. In addition, common autoimmune diseases such as thyroid associated ophthalmopathy, Sjogren's disease, multiple sclerosis and Behcet's disease may develop in MG patients, further supporting ophthalmic complications. In generalized MG, the incidence of autoimmune disease is higher and more variable than that in ocular MG patients; therefore, generalized MG patients should be screened for ophthalmic complications on a regular basis. In MG patients with long-term steroid use, glaucoma may develop due to optic nerve damage and can lead to irreversible visual field defects. Since loss of vision is often only recognized when the disease is advanced, early detection of glaucoma is very important in a background of MG. Long term steroid treatment in MG patients may also cause complications of posterior polar cataract and central serous chorioretinopathy, effects which must be taken into careful consideration.
Notes
No potential conflict of interest relevant to this article was reported.