Introduction
Inherited retinal dystrophies (IRDs) constitute a group of phenotypically and genetically heterogeneous retinal diseases associated with the progressive loss of vision [1]. Pathogenic variants in more than 270 various genes have been implicated in IRD etiology [2]. RPE65 encodes the 65-kDa isomerase of the visual cycle, which converts all-trans-retinyl esters to 11-cis-retinol. The mutated product of this gene in the retinal pigment epithelium (RPE) affects the visual cycle, which leads to a progressive loss of photo-receptors. RPE65-associated retinal dystrophy can present with various forms of IRDs, such as Leber congenital amaurosis (LCA), early-onset severe retinal dystrophy (EOS-RD), and early severe retinitis pigmentosa (RP), depending on the effect of causative variants [1]. Because the clinical presentation of RPE65-associated retinal dystrophy is highly heterogeneous, accurate diagnosis is often challenging at the time of presentation [3].
Given the circumstances mentioned above and considering the growing availability of gene therapies, a molecular diagnosis is an essential component in evaluating patients with suspected RPE65-associated retinal dystrophy. In this paper, we review available data on the epidemiology, molecular genetics and clinical characteristics of RPE65-associated retinal dystrophy.
Epidemiology of RPE65-associated Retinal Dystrophy
The prevalence of LCA is estimated at 1.20 to 2.37 per 100,000 [4-6]. The proportions of RPE65-associated LCA varied considerably in various countries, from 1.26% to up to 95% in clinically diagnosed patients [2,7-42] and from 3.95% to 40% among those with a molecular diagnosis of IRD [2,25,27,37,42-46]. The proportion of RPE65 gene mutations in LCA families worldwide was estimated at 6.15% [47], but the percentages of the mutation-carrying families from various countries differed substantially, from 1% to 18.18% in clinically diagnosed cases [48-52] and from 7.14% to 13.64% in molecularly diagnosed ones [2,53,54]. The prevalence of RP worldwide was reported at 11.09 to 47.62 per 100,000 [5,55-68]. The proportions of RPE65 mutations in clinically and molecularly diagnosed patients with RP varied, from 0.23% to 10.34% [8,11,13,23,25,69-73] and from 3% to 21.43% [25,71,73,74], respectively. Meanwhile, the proportion of RPE65 mutations in RP families ranged from 0.43% to 3.95% [75-77].
In a survey conducted by our group in Korea in May 2022, biallelic RPE65 mutations were found in six out of 2,140 patients (0.28%), including 109 LCA and 2,031 RP patients (unpublished data). In other countries, the proportion of RPE65-associated retinal dystrophy ranged from 0.60% to 20% [2,6,7,12,13,25,43,48,49,67,78-93]. The results of those studies are summarized in Table 1 [2,6-46,48-54,67,69-93].
To summarize, available evidence shows that RPE65-associated retinal dystrophy can occur in any population, regardless of geography and ethnicity. The global prevalence of RPE65 mutations appears to be higher among LCA patients (about 5-10%) than among RP patients (about <5%), which might be associated with greater genetic heterogeneity of RP compared with LCA. The proportions of RPE65-associated retinal dystrophies among molecularly diagnosed cases were shown to vary considerably depending on the study type and patient ethnicity, and so was the prevalence of LCA and RP. Those discrepancies might be associated with the influence of multiple confounding factors, such as differences in study designs and inclusion criteria for molecular analyses, nonstandardized diagnostic terms and poorly defined diagnostic criteria, and high consanguinity of some examined populations, to mention a few. All those issues need to be addressed in future studies to obtain an unbiased insight into the epidemiology of RPE65-associated retinal dystrophy.
Molecular Background of RPE65-associated Retinal Dystrophy
RPE65 gene is located on chromosome 1 (1p31.3), spanning over 20 kb and including 14 coding exons [94,95]. Human RPE65 encodes the RPE-specific 65 kDa protein (RPE65), currently referred to as retinoid isomerohydrolase RPE65 [95,96]. RPE65 is a highly conserved protein consisting of 533 amino acids, expressed at high levels exclusively in the RPE [95,96]. The amino acid sequence of RPE65 shares 98%, 94%, 82%, and 74% identity with bovine, mouse, frog, and zebrafish, respectively [97]. RPE65 plays a role in the visual cycle, a complex process during which light entering the eye is converted into electrical signals. Reaching photosensitive pigments in the retina, light causes the conversion of 11-cis-retinal to all-trans-retinal. RPE65 reconverts all-trans-retinyl ester to 11-cis-retinol, thus enabling a new photoisomerization event [95]. In the absence of RPE65, the level of 11-cis-retinol decreases and retinyl esters accumulate within the RPE [95].
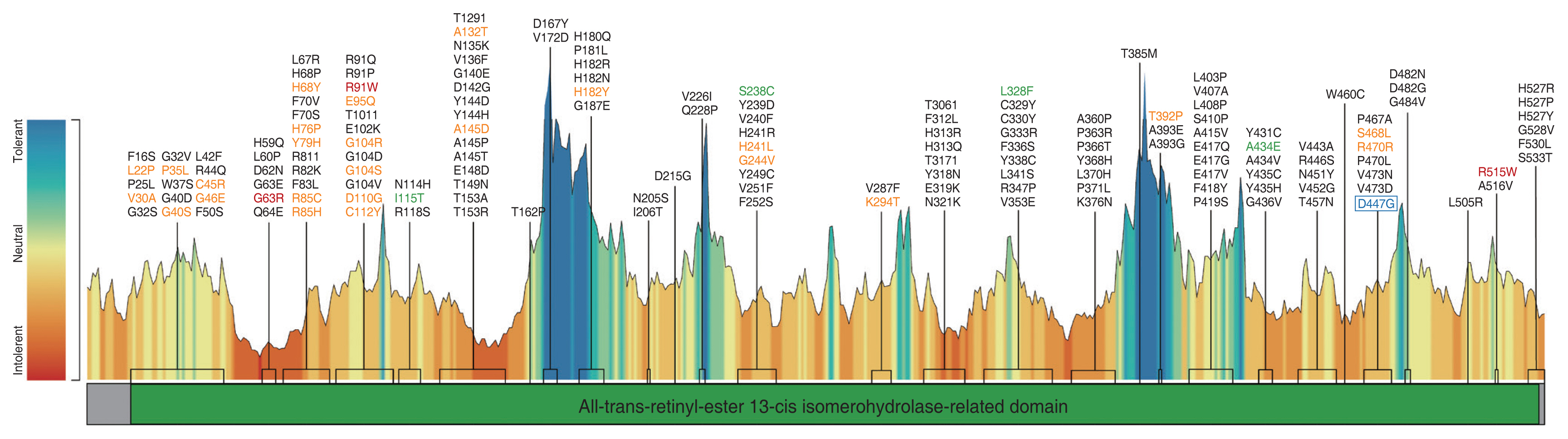
As of March 7, 2023, 776 variants of the RPE65 gene are listed in the ClinVar database [98], among them 162 pathogenic, 65 likely pathogenic, and 231 variants of uncertain significance (VUS). Most of the reported variations (n = 671) are single-nucleotide variants (SNV) [98]. Another database, the Leiden Open Variation Database (Leiden University Medical Center; https://www.lovd.nl/3.0/home/), lists a total of 364 RPE65 variations, among them 280 pathogenic or likely pathogenic and 60 VUS [99]. In Human Gene Mutation Database Professional (Institute of Medical Genetics, Cardiff University; https://www.hgmd.cf.ac.uk/ac/index.php/) [100], a total of 292 disease-causing variants were reported. Among them were 194 SNVs, 36 splicing substitutions, 58 small indels, three gross deletions, and one complex rearrangement [100]. Finally, the Genome Aggregation Database (https://gnomad.broadinstitute.org/) contains 120 synonymous SNVs of RPE65, 284 missense SNVs, and 24 SNVs marked as “putative loss-of-function” [101]. The distribution of reported missense variants associated with LCA, RP, and fundus albipunctatus is shown in the tolerance landscape generated using the Metadome web server (Fig. 1) [102].
While previous studies found no link between the presence of a specific RPE65 genotype and phenotypic and clinical characteristics of RPE65-associated retinal dystrophy [103,104], recent evidence suggests that a relationship may exist between the mutation type and the age of disease onset [48]. Specifically, patients with two missense alleles were shown to develop the disease later (≥1 year of age) than those with one or two truncating variants (<1 year of age) [48].
Genetic Testing for RPE65-associated Retinal Dystrophy
Nowadays, the underlying genetic cause of IRD can be detected in up to 76% of patients [105]. Genetic testing for RPE65-associated retinal dystrophy can be performed by Sanger sequencing, especially when next-generation sequencing (NGS) is not available. It has been postulated that patients with suspected RPE65-associated retinal dystrophy, especially those with VUS, should also be tested for mutations in other IRD genes [105,106]. An ocular NGS panel containing sufficient IRD genes is highly recommended in such cases.
To provide the result of adequate quality, a diagnostic laboratory must be certified, equipped with state-of-the-art devices, and employ personnel experienced with modern technologies, such as Sanger sequencing, NGS, exome sequencing (ES), and multiplex ligation probe amplification (MLPA). Choosing the testing strategy, one should consider its cost, turnaround time, coverage, and data storage capacities. Most commonly, the first-line technologies include a targeted NGS panel, clinical ES or ES followed by targeted in silico analysis restricted to genes implicated in IRDs. The remaining genes are analyzed only when no ultimate diagnosis has been obtained with the ocular panel.
Genome sequencing (GS) can be performed in patients in whom no pathogenic variants were found on targeted testing although clinical phenotype suggests otherwise or those unresolved by ES [107,108]. Especially if RPE65-associated retinal dystrophy is clinically suspected, but only a monoallelic variant in RPE65 is detected, one should assume the other allele carries a deep intronic or structural variant. In such case, further diagnostic workup, including GS, MLPA, and microarray-based comparative genome hybridization, should be considered.
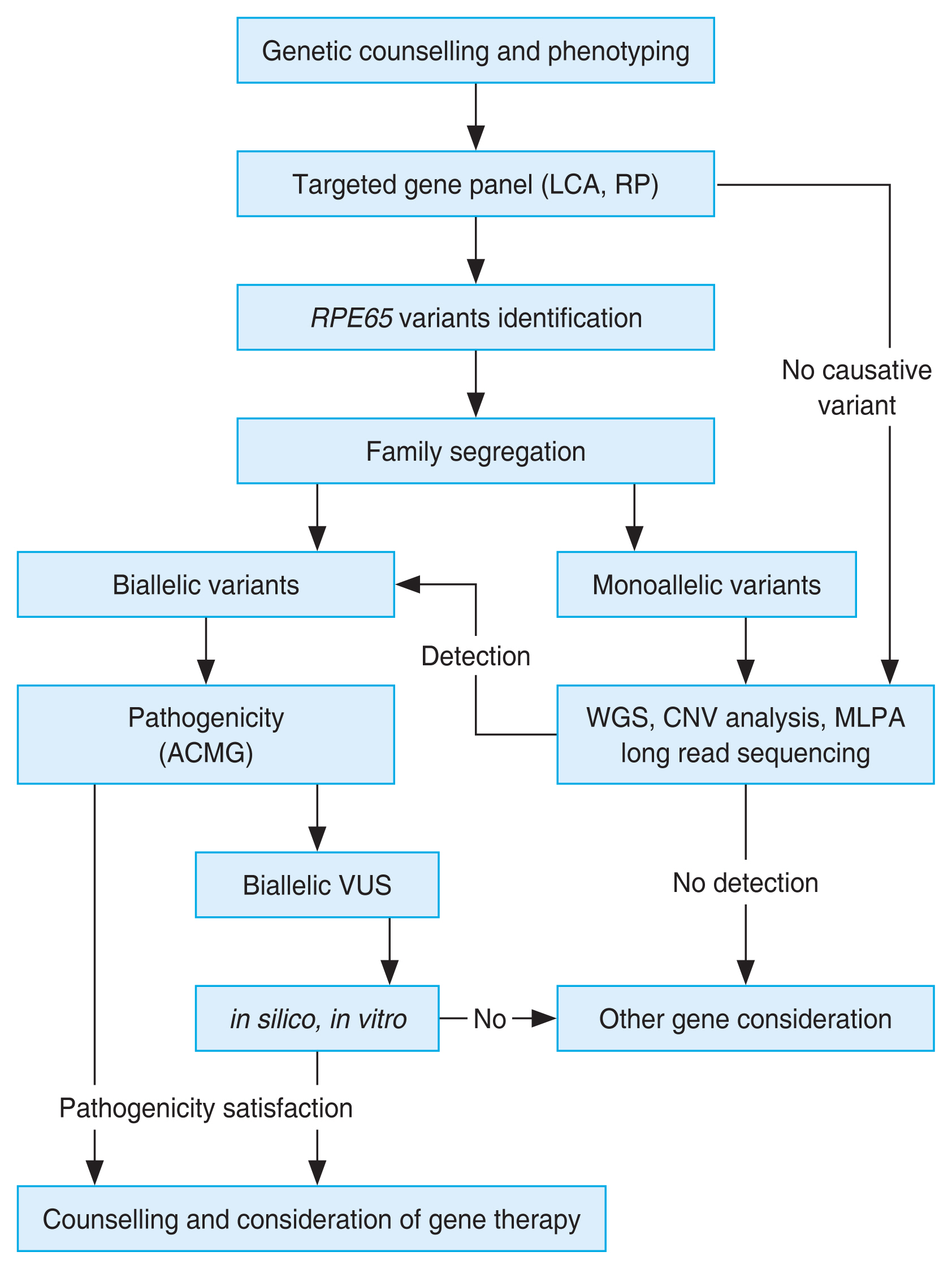
One proposed approach to genetic workup in patients with suspected RPE65-associated retinal dystrophy, including all the technologies discussed above [109], is presented schematically in Fig. 2.
Theoretically, the occurrence of VUS among the RPE65 variants might constitute an obstacle in determining the eligibility for gene therapy in IRD [110]. Importantly, however, no correlation between variant subtype or the American College of Medical Genetics and Genomics classification and treatment response was found in one previous study [111]. In that study, at least one VUS was identified in seven out of 29 patients with a confirmed genetic diagnosis of biallelic RPE65 gene variants; none of those patients, including three persons with two VUS, did fail to respond to gene supplementation therapy [111].
If a VUS in the RPE65 gene has been identified, its pathogenicity can be determined with various methods, namely in extended segregation studies, through the evaluation of the phenotype, in silico tools to predict protein conservation and functionality, and in vitro functional studies.
The problem of the potential involvement of compound heterozygous variants in IRD etiology may be addressed by the segregation analysis of the proband’s parents. Theoretically, a patient may be exempted from the segregation analysis when he/she was shown to be homozygous for a known variant, and the result of genetic testing is consistent with a phenotypic presentation. Nevertheless, segregation analysis of the apparently “homozygous” variants is always recommended to obtain a better insight into the genotype, as the patient with such variants may show copy number variations leading to loss of heterozygosity [112,113], uniparental isodisomy [114], or true homozygosity due to the existence of genetic isolates [115]. Moreover, some patients may carry two variants in cis rather than in trans. To distinguish whether two variants are in cis or trans, segregation or trio analysis using parental DNA should be performed. If it is difficult to use the parents’ samples, haplotype resolutions using long reads are an approach of choice to assign genetic variants to the homologous paternal and maternal chromosomes [116].
Importantly, dominantly inherited RPE65-associated retinal dystrophy has been reported as well, such as c.1430A>G:p.(Asp477Gly) mutation; however, the efficacy of gene augmentation therapy in the heterozygous dominant-negative RPE65 variant has not been verified [117,118].
Based on the available evidence and our clinical experiences, the chance of detecting the RPE65 variant in Korean patients with LCA/RP is presumed lower than in Western populations [38,119]. However, the rarity of the disease does not preclude its treatability. Besides, Korea provides good medical accessibility, and the national health insurance covers NGS-panel sequencing for IRDs, including LCA and RP. Therefore, a more proactive attempt in making the molecular diagnosis is recommended in patients suspected to carry RPE65-associated diseases, not to miss any case that could be treated.
Even though most of the multigene testing panels for IRD include RPE65, each laboratory uses different testing panels that offer an array of various target genes [120]. Considering this, one should check if the testing panel includes RPE65 containing 14 exons that can induce enough sequencing depth from an exon-intron junction.
When a new NGS panel is constructed, or the existing gene testing panel is to be updated, the following issues should be considered. First, the panel should include the deep intronic variants and the identified splicing variants of RPE65, especially the variant that could be easily missed as it is 5 to 10 base pairs away from the intron-exon junction. Secondly, copy number variants analysis should be routinely performed and implemented in the report. Thirdly, whole GS should be additionally considered to detect structural variation, intronic variant, or other variants when only one allele of RPE65 harbors the variant without other causative genes.
Clinical Diagnosis and Characteristics in RPE65-associated Retinal Dystrophy
Genetic testing is essential in IRDs in order to establish a definitive diagnosis. However, it is important to thoroughly review the clinical features before performing genetic testing. Several diagnostic modalities, including fundus photograph, electroretinogram (ERG), optical coherence tomography (OCT), and fundus autof luorescence (FAF) imaging, can be used to diagnose and differentiate IRDs. However, there are limitations in young children because they do not cooperate well with comprehensive eye examinations. Also, in patients with nystagmus, obtaining high-quality retinal images is difficult. In the case of ERG, it usually requires sedation, and the waveform of ERG may appear small, even in normal children. Due to the limitations of these tests, other diseases can be misdiagnosed as LCA/EOSRD [121].
LCA/EOSRD is often associated with several systemic diseases [122]. Senior-Løken syndrome, Joubert syndrome, Bardet-Biedl syndrome, Zellweger syndrome, Refsum disease, Alström syndrome and Walker-Warburg syndrome are diseases that cause retinal degeneration along with systemic involvement [123]. However, no case of RPE65-associated retinal dystrophy related to a systemic disease has been reported thus far. Consequently, if a patient has both a systemic disease and retinal degeneration, RPE65-retinal dystrophy could theoretically be ruled out. However, it cannot be excluded that the two conditions occur concomitantly in a single patient without a causal relationship.
Most patients with RPE65-associated retinal dystrophy present with early-onset visual loss or difficulty in night vision, frequently accompanied by nystagmus or wandering eye movement. Oculodigital signs are not typically reported in this group of LCAs [124]. Some patients with RPE65-associated retinal dystrophy do not present with nystagmus. The presence of nystagmus is considered an important feature distinguishing between LCA and EOS-RD. In patients with RPE65-associated LCA, infantile-onset nystagmus is almost always present.
In one study, the best-corrected visual acuity (BCVA) in patients with RPE65-associated LCA ranged between 20 / 32 and no light perception, with a median of 20 / 225 [125]. According to previous studies, BCVA in RPE65-associated EOSRD was 20 / 50, whereas median BCVA in RPE65-associated RP amounted to 20 / 80 [124]. The BCVA in RPE65-associated LCA/EOSRD was typically between 20 / 400 and 20 / 200 and was relatively well maintained up to 3 to 18 years of age. In an Italian cohort, more than half of patients presented with nystagmus, and photophobia was found in 46.5% of the cases. Because VA is relatively preserved during early infancy in the subtype of RPE65-associated EOSRD or late-onset RPE65-associated RP, nystagmus may be absent in these disorders.
The symptoms of RPE65-associated retinal dystrophy typically appear between birth and the age of 5 years. By the time the patients reach school age, they usually present with profound nyctalopia. In patients with RPE65-associated retinal dystrophy, VA can deteriorate severely and progress to legal blindness by the age of 20. Although no strong correlations between genotype and phenotype had been reported [126], Lopez-Rodriguez et al. [48] reported that 60% of patients with a missense or missense genotype showed symptoms before or during the first year of age.
Variable degrees of refractive errors had been reported in patients with RPE65-associated retinal dystrophy. According to Chung et al. [3], the majority of refractive errors found in 64 patients were myopic (57.4%) and hypermetropic errors (39%); only four patients (6.3%) showed emmetropia. Within the myopia group, about 50% of patients presented with a low degree of myopia (<-3 diopters). In an Italian cohort, about 50% and 25% of patients had a myopic and hypermetropic spherical equivalent, respectively [127]. In other studies, patients with RPE65-associated retinal dystrophy often presented with high hyperopia [49,128].
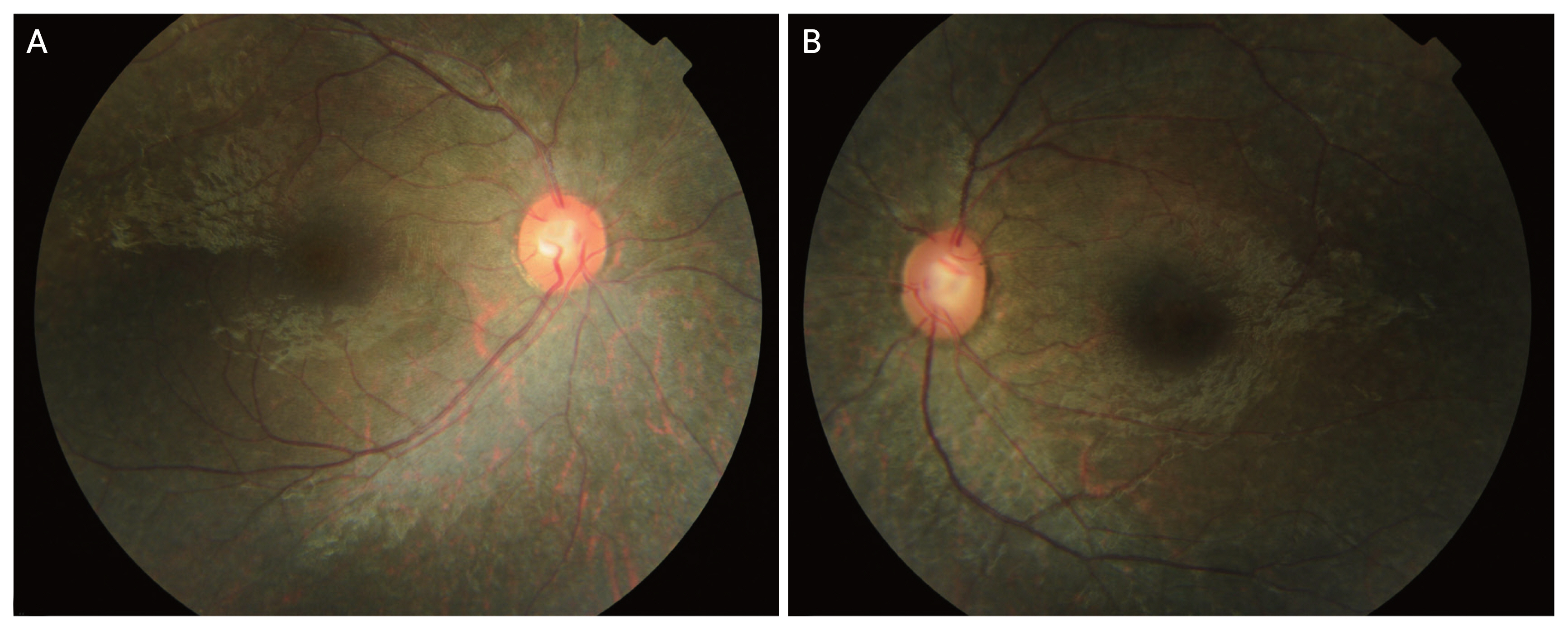
The fundus abnormalities in patients with RPE65-associated retinal dystrophy can be quite variable, and typically a normal-appearing fundus is observed in infancy [106]. Therefore, RPE65-associated retinal dystrophy can be frequently missed or misdiagnosed in early infancy. In early childhood, hypopigmentation and fine granularity may be found in the retina [129]. Paunescu et al. [130] noted that the hypopigmentation had a characteristic peripapillary distribution around and inferiorly to the optic disc (Figs. 3A, 3B, 4A, 4B). In some cases, the retina may show fine white dots, corresponding to an abnormal accumulation of retinyl esters. These small round white deposits reside within the RPE and can be detected by OCT; the deposits may wax and wane and eventually fade with time [131]. The white-yellowish deposits are replaced by RPE dropout and atrophy, with more substantial mottling and pigment clumping occurring over time. Diffuse granular retinal dystrophy or salt-and-pepper retinal dystrophy can be seen in early teen ages (Figs. 3, 4). In their 30s or 40s, as RPE dystrophy with fine pigment clumping and dispersion progresses, typical RP with bony spicule pigmentation becomes more prominent. In other words, patients with typical pigmentary changes are mostly older than 35 years. In addition, fundus albipunctatus (small round white dot deposits in the midperipheral retina) can be seen in patients with biallelic RPE65 variants [49,132]. Therefore, RPE65-associated retinal dystrophy should be considered an underlying condition in patients with fundus albipunctatus.
The waveforms of ERG are undetectable or severely attenuated in all patients with RPE65-associated retinal dystrophy, even if fundus examination seems normal. When the onset of symptoms is late (i.e., between 1 and 5 years of age), some residual rod activity on dark-adapted ERGs or cone function on light-adapted ERGs can be detected.
The OCT findings show diffuse loss of the ellipsoid zone with relatively well-preserved central foveal architecture (Figs. 5A, 5B, 6A-6F). The outer nuclear layers are also thinned throughout the entire retinal area. On a histopathological examination, decreased inner nuclear layer, thinning of the outer nuclear layer, and normal ganglion cell layer can be observed in RPE65-associated retinal dystrophy [133]. In patients in their 20s or 30s, cystoid macular edema, macular hole, epiretinal membrane or vitreomacular tractions may be found on OCT scans. A minority of patients have signs of RPE atrophy [127]. The thickness of the foveal outer nuclear layer was shown to decrease with age [134,135].
FAF can detect autofluorescent materials (e.g., lipofuscin and melanin) within the RPE and choroid [124]. Short-wavelength autofluorescence imaging may show extinguished signals in both central macular areas and the entire retinal periphery (Fig. 7A, 7B). However, near-infrared FAF imaging can demonstrate a relatively preserved autofluorescence in the central fovea area (Fig. 8A, 8B). Therefore, a relatively preserved central retinal structure on OCT and near-infrared FAF in the presence of very abnormal or undetectable short-wavelength FAF signals could be a clinical marker of this specific molecular subtype of RPE65-associated retinal dystrophy [136]. According to Kumaran et al. [137], FAF was observed in 46% of patients with RPE65-associated retinal dystrophy. In contrast, Lorenz et al. [136] showed that FAF is nearly absent or minimal in patients with this condition.
IRD can be easily misdiagnosed as functional visual loss or optic nerve disease, especially at a young age. When clinical signs indicate a possibility of IRD, the clinician should not exclude this condition solely based on normal findings in fundus examination, and molecular examination should be included in the diagnostic plan, even without ERG or OCT. We again emphasize that early diagnosis is of utmost importance in managing IRD.
In summary, the clinical diagnosis of RPE65-associated retinal dystrophy based on a single diagnostic clue can be challenging, given that clinical features and ophthalmological findings can vary considerably. Clinical symptoms and ophthalmological findings in RPE65-associated retinal dystrophy can vary from nearly normal to severe abnormalities [106]. Hence, careful observation is required to diagnose and suspect RPE65-associated retinal dystrophy because clinical diagnosis should precede genetic investigations. Ultimately, genetic testing is required to confirm the disease.
Voretigene Neparvovec
Voretigene neparvovec (VN), the first approved gene therapy for a genetic disease, is a recombinant adeno-associated virus 2 vector containing human RPE65 complementary DNA that enables RPE cells to produce the retinoid isomerohydrolase RPE65. After its efficacy and safety were ultimately confirmed in an open-label, randomized, controlled phase 3 trial conducted at two centers in the United States, VN has been authorized for gene augmentation therapy in RPE65-associated retinal dystrophy [138].
The trial mentioned above [138] was conducted at two centers in the United States. The study included patients with confirmed genetic diagnosis of RPE65-associated inherited retinal dystrophy, age ≥3 years, bilateral BCVA of no more than 20 / 60 and/or visual field <20° in any meridian, and sufficient viable retina. A total of 31 patients satisfying the criteria mentioned above were enrolled and randomly assigned (2:1) to the intervention (n = 21) or control arm (n = 10). Of this number, 29 patients eventually entered the study, among them 20 from the intervention arm and nine from the control arm. Patients from the intervention arm received 1.5 × 1011 vector genomes (vg) of VN in a total volume of 0.3 mL as a subretinal injection to the first eye, followed by the injection to the second eye 6 to 18 days later. No sham procedure was performed in patients from the control arm, but they underwent the same efficacy testing as those from the intervention arm. After a 1-year follow-up, mean bilateral change in a standardized multiluminance mobility testing (MLMT) score was significantly higher in the intervention arm than in the control arm (1.8 ± 1.1 vs. 0.2 ± 1.0, p = 0.0013), with 13 participants (65%) from the intervention arm but none from the control arm passing the test at the lowest possible luminance level (1 lux). While the mean white light full-field stimulus threshold (FST) in the intervention arm improved substantially, by more than 2 log cd · sec/m2 by day 30 postinjection, and then remained stable till the end of the follow-up at 1 year, no clinically meaningful change in mean white light FST was observed in the control arm (between-group difference −2.11 log cd · sec/m2, p = 0.0004).
Meanwhile, some benefits in terms of BCVA were observed as well, with a 9.0 and 1.6 letter improvement in the intervention and control arm, respectively, when averaged for both eyes (difference 7.4 letters, p = 0.0469). As per visual field testing, the intervention arm performed significantly better in terms of the mean sum total degrees of Goldman visual field (GVF; III4e) and macula sensitivity threshold on Humphrey visual field testing, whereas no significant between-group difference was observed in Humphrey foveal sensitivity threshold. No product-related adverse events or deleterious immune responses were recorded. The majority of ocular adverse events (most commonly eye inflammation, elevated intraocular pressure, and cataract) were mild in severity. Two patients experienced serious adverse events unrelated to the study product [138].
Eligibility for VN Treatment
VN is indicated for the treatment of patients with confirmed biallelic RPE65-associated retinal dystrophy. Patients must have viable retinal cells as determined by the treating physician. However, clear criteria to determine the presence of viable retinal cells have not been provided thus far. The criteria for the presence of sufficient viable retinal cells were defined as follows in the phase 3 trial [138]. The patient must have either the following: (1) an area of the retina within the posterior pole of >100 μm thickness shown on OCT; (2) ≥3 disc areas of the retina without atrophy or pigmentary degeneration within the posterior pole; or (3) remaining visual field within 30° of fixation as measured by a III4e isopter or equivalent [138]. Until now, those inclusion criteria appear to be the most helpful for the treating physician to determine the presence of viable retinal cells.
Aside from the presence of viable retinal cells, residual visual function and the results of anatomical evaluation using multimodal imaging should also be considered when assessing eligibility for VN treatment. The phase 3 clinical trial included patients with VA worse than 20 / 60 (both eyes) and/or a visual field of less than 20° in any meridian as measured by a III4e isopter or equivalent (both eyes) [138]. Although the potential benefit of gene therapy in patients without sufficient viable retinal cells can be limited, we have found no rationale to set a minimum VA as an eligibility criterion for the presence of sufficient viable retinal cells. Published evidence suggests that even patients with very low VA (e.g., hand movement [HM] or count fingers VA) may show some improvement in night vision and/or VA after VN treatment. In two out of three first patients treated with RPE65 gene therapy, baseline VA was HM. Nevertheless, those two patients showed evidence of improvement in retinal function [139]. Also, in the phase 1 dose-escalating study, all three patients with poor baseline visual acuity (2.0 logarithm of the minimum angle of resolution [logMAR] or worse) showed an improvement in light sensitivity [140]. Finally, in the phase 3 trial of VN, one patient with poor baseline VA showed some improvement in the visual field and light sensitivity [138].
The phase 3 clinical trial of VN included patients aged 3 years or older [138]. According to the prescription information, the use of VN in infants under 12 months of age is not recommended; this is warranted by the risk of potential dilution or loss of VN after its administration due to the active retinal cell proliferation occurring in this age group. Although early treatment is recommended to prevent further deterioration of visual function, treating patients aged 12 to 36 months should be undertaken cautiously, given limited published evidence for this age group, more challenging surgical procedures and inability to evaluate pretreatment and posttreatment VA accurately. Moreover, the authors of a recently published case series reported the presence of subretinal deposits after VN administration in young patients with RPE65-associated retinal dystrophy, pointing to potential safety implications [141].
In addition, we have not found any rationale to set a maximum eligibility age for VN treatment. Most patients diagnosed with RPE65-associated IRD, including the two patients treated with VN in Korea, were in their late 20s or older, which is generally an older age group than the populations included in the regulatory trials of VN or real-world studies published so far. Based on our experience, although limited, we strongly recommend genetic testing for all those under 30 years of age, even if a patient presents with RP at an older age. It also needs to be stressed that one exclusion criterion applied in phase 3 clinical trial of VN, the use of retinoid compounds or precursors [138], should not disqualify patients from the treatment in real-world clinical practice.
Retinal Gene Therapy with VN
Subretinal gene therapy with VN should only be performed in specialized centers possessing appropriate equipment for proper storage and preparation of the injection solutions and having adequate experience with macular surgery.
In line with the manufacturer’s specifications, 0.3 mL of the vector suspension (dose, 1.5 × 1011 vg) should be administered via retinal injection using pars plana vitrectomy (e.g., 23- or 25-gauge), followed by air tamponade. Treatment of the fellow eye should be performed within a short period of time but at least 6 days apart. While VN is stable in an aqueous solution, it should be stored at temperatures below −65°C until use, and the cold chain should be maintained and documented appropriately. VN can be prepared for administration as early as 4 hours before the treatment. The preparation procedure should be carried out using an aseptic technique and under sterile conditions in class II vertical laminar flow biological safety cabinet. The instruments and materials needed to prepare VN should be available in the pharmacy department of the treatment center. The personnel should be trained in handling biosafety level 1 agents and using a safety cabinet. To ensure compliance with the manufacturer’s regulations and all safety requirements, standard operating procedures are recommended to be developed and implemented for the reconstitution and disposal of waste products. The sterility of the virus suspension needs to be ensured during preparation and transport to the operating room.
The surgical center should be stocked with all instruments and materials required for VN administration (e.g., a 38- or 41-gauge subretinal cannula). The entire surgical team should be trained in handling biosafety level 1 agents. The surgeon should have experience in subretinal surgery and vitreoretinal surgery in young patients. Ideally, the surgeon should be experienced in the surgical management of the eye in patients with retinal dystrophies. The recommended site of injection should be located along the superior vascular arcade, at least 2 mm from the fovea center; the injection should be performed in such a way that all pathologic areas, such as dense atrophy or intraretinal pigment migration, are avoided. A small amount of the virus suspension should be slowly injected until an initial subretinal bleb is observed. Then the remaining volume should be injected slowly until a total of 0.3 mL is delivered.
The use of an operating microscope with intraoperative OCT may be helpful, along with a vitrectomy system that enables the surgeon to control the rate of vector injection. An intraoperative OCT allows the surgeon to confirm that the vector is being administered into subretinal space and to objectively identify the localization and extent of subretinal injection. Subsequent fluid-air exchange is recommended to eliminate potential virus particles from the vitreous cavity. Care should be taken not to aspirate the material near the retinotomy site.
Supine head positioning of the patient should be initiated immediately and maintained for the first 24 hours of the postoperative period. Disposal of the remained virus suspension and backup syringe should be carried out according to local biosafety guidelines for handling and disposal of the product.
Prednisolone (1 mg/kg body weight per day, but no more than 40 mg/day) should be administered for 7 days in total (starting 3 days before VN administration), and then, the maximum dose should be tapered over 10 days. The same corticosteroid dosing regimen applies to administering VN to the second eye. If the second eye is to be treated shortly after the first one, tapering should be discontinued, and the original corticosteroid regimen should be initiated as described above.
Follow-up measurements are recommended to be obtained 1, 3, 6, and 12 months after the treatment and yearly thereafter, for about up to 5 years, as required by local regulations. Visual functions that showed a significant improvement in clinical trials of VN included orientation and mobility at low light levels (MLMT) and retinal sensitivity as measured by the FST testing [138]. MLMT showed a good correlation with FST and macular threshold in Humphrey visual field test. As for the FST, a change that exceeds the test-retest variability threshold of 0.3 [142] should be considered clinically beneficial for an individual patient. Any improvement, even lower than the aforementioned threshold, can be a meaningful change considering the progressive nature of the disease. In the phase 3 trial of VN [138], improvements in visual function were most pronounced between 2 weeks and 6 months posttreatment and, in some cases, could still be demonstrated at 5 years [143]. Based on this evidence, FST should be implemented as an outcome measurement test, and other outcome measures, such as BCVA, fundoscopy, OCT, and visual field testing, can be included as supplementary information in cooperating patients during follow-up. Additional valuable information can be obtained from MLMT, fundus-controlled perimetry, FAF, ERG, visual evoked potential, and eye movement recordings.
Real-world Evidence
As mentioned before, the efficacy and safety of VN were verified in an open-label, randomized, controlled phase 3 trial conducted in the United States [138]. Based on those findings, in 2017, the treatment was authorized in the United States, followed by approval in other countries, including the European Union member states (2018), Australia (2020), and Korea (2021).
After VN had been approved, a substantial body of real-world evidence was gathered concerning the efficacy and safety of the treatment. The largest two real-world studies of VN have been conducted in the United States, including 77 eyes from 41 patients and 27 eyes from 15 patients, respectively [144,145]. Some additional evidence regarding the real-world outcomes in VN-treated patients originates from individual case reports and small case series, including one case report of a Korean patient [84,141,146-152].
Additionally, a real-world global post-authorization study, PERCEIVE, is currently ongoing [153]. The enrollment in the study started in 2019 and will continue until 2024, with each patient being followed up for 5 years. A total of 106 patients from 15 non-US countries have been enrolled till August 31, 2021 (data cutoff), with 103 deemed eligible for the study. At the time of data cutoff, VN was administered to 183 eyes. The study patients have been followed up for a mean period of 0.8 ± 0.64 years (maximum, 2.3 years) [153].
Real-world effectiveness of VN treatment
1) Full-field stimulus threshold
The primary outcome measure of VN treatment effectiveness in the real-world studies mentioned above was FST. At the time of data cutoff, at least 6-month follow-up data were available for 42 eyes of patients enrolled in the PERCEIVE study [153]. A rapid increase in white light sensitivity was observed in this subset, from −4.56 dB at baseline to −16.59 dB at month 1 and −18.24 dB at month 6 posttreatment [153]. A similar degree of white light FST improvement was also documented in the multicenter retrospective analysis conducted in the United States [144]. In that study, mean white light FST improved significantly, from 0.6 ± 3.7 dB at baseline to −20.5 ± 19.2 dB at final testing (mean change, 21.1 ± 16.6 dB; p < 0.001) [144]. A significant improvement in FST, by 2.1 log cd · sec/m2 on average, was also documented at 6 to 12 months posttreatment in all 13 eyes included in the single-center retrospective analysis conducted in the United States [145]. This evidence of VN effectiveness in terms of light sensitivity improvement is also supported by data from small case series and individual case reports [147-149]. Among the patients included in those reports was a 30-year-old Korean patient who received a VN injection to the left eye. White light FST in the patient improved from −3.13 dB before treatment to −23.4 and −29.8 dB at 1 and 3 months, respectively [148].
2) Visual field
In the multicenter retrospective analysis conducted in the United States [144], GVF outcomes at the baseline and at least 2 months after VN injection were available for 16 eyes. Of that number, 11 eyes showed an expansion of the visual field area, whereas a decrease was observed in another five eyes. The mean area for V4e stimuli increased, from 4,767 ± 4,518.7 deg2 at baseline to 5,701 ± 2,916 deg2 at final testing, but the change did not turn out to be statistically significant (mean change, 934 ± 2,916.4 deg2; p = 0.34) [144]. Meanwhile, a significant change in GVF was observed in all 13 eyes included in the single-center US-based analysis, with a 221 sum degrees increase from the baseline (p < 0.001) [145]. A significant enlargement of the visual field was also documented in small case series and individual case reports, among them one concerning a Korean patient [146-149].
3) Visual acuity
No clinically meaningful change in BCVA was observed over time in the interim analysis of the PERCEIVE study results, as well as in the multicenter retrospective analysis of VN treatment outcomes in three US-based centers [144,153]. In contrast, a significant improvement of BCVA, from 0.98 logMAR at baseline to 0.83 logMAR at the last follow-up (p < 0.001), was documented in the retrospective analysis of pediatric patients treated at a single center in the United States [145]. Equally inconclusive are the data from small case series and individual case reports [146-149], with some studies showing an improvement in visual acuity and others not. For example, in a Korean adult patient, VA in the treated eye improved from 20 / 200 before treatment to 20 / 400 at 1 month and 20 / 100 at 3 months [148]. It needs to be stressed that while VA improvement is unlikely, given the mechanism of VN action, available evidence suggests that some patients might benefit from the treatment also with regards to this parameter.
Real-world safety data
The spectrum of posttreatment complications documented in available real-world studies [144,145,153] was generally similar to that reported in the phase 3 trial of VN [138]. However, a few previously unreported complications that deserve special attention were identified as well.
French authors reported a rare complication, macular fold formation, in a 23-year-old patient [150]. On the 1st day after a standardized VN injection, the patient reported a loss of central vision in the treated eye, and a large macular fold involving the fovea was found on fundoscopy. A surgical revision resulted in a complete regression of the retinal fold, but a substantial alteration persisted in the macular profile. While the patient demonstrated light sensitivity improvement at 1-month follow-up, visual acuity in the treated eye still remained low. The complication is believed to result from the conflict between the air bubble created inadvertently during retinal detachment and another subretinal bubble caused by the VN injection. Additionally, the patient did not follow the recommended strict supine position regimen within the initial 24 hours postprocedure [150]. On the interim analysis of the PERCEIVE study results, the most common complication was chorioretinal atrophy at the injection site and/or other location, found in 19 eyes from 13 patients with a mean age of 20.5 years (range, 9-33 years) [153]. A similar complication, not documented in the phase 3 trial [138], was also found in seven eyes from patients treated in three US-based centers [144]. The most recent evidence of chorioretinal atrophy after VN injection originates from a multi-center retrospective review of 18 eyes from 10 patients with LCA [154]. The patients developed progressive perifoveal chorioretinal atrophy after the procedure, which progressively enlarged over time. However, there was no significant change in VA. The study also found that all eyes with reliable GVF demonstrated improvement, but 23.1% of them presented with paracentral scotomas related to the atrophy. Unfortunately, the review included solely the eyes that developed the complication, and hence, did not provide an insight into the overall incidence of perifoveal chorioretinal atrophy or differences, if any, between eyes with the atrophy and without [154].
Authors from another US center reported a series of three pediatric patients aged 22 months to 5 years who developed subretinal deposits within a week from VN injection [141]. The deposits, distant from the original bleb location, resolved gradually within 4 months to 2 years posttreatment. As emphasized by the authors, all patients who developed subretinal deposits were young children, two of whom would not have met the age cutoff for the phase 3 clinical trial [138]. The authors speculated that the deposits, probably representing a transient immune response to the viral vector, might have been a consequence of poor compliance with supine positioning after injection and the resultant overexposure of ocular tissues to the vector [141].
Other real-world findings to be considered
Authors from Saudi Arabia presented a case series of three siblings with unusual late presentation of RPE65-associated retinal dystrophy, diagnosed at the age of over 30 years [84]. Despite the advanced age at the diagnosis, all patients were deemed eligible for gene therapy, as their central retinal thickness exceeded 100 microns on repeated examinations. Thus, the authors postulated that late-presentation RPE65-associated retinal dystrophy might constitute an indication for VN treatment providing an adequate function of the remaining retina and structural preservation. A therapeutic window for intervention in such a group of patients is yet to be determined [84].
A group of authors from the United States proposed a new surgical technique dedicated to pediatric patients, a group in which the injection of VN is considered particularly challenging [151].
German authors tested two alternative methods to assess the VN treatment outcome, dark-adapted chromatic parameter (DAC) with a new shortened protocol and chromatic pupil campimetry (CPC). According to the authors of that study, CPC and DAC present new and fast ways to assess functional changes in retinotopic maps of rod and cone function, measuring complementary aspects of retinal function which are not determinable with other routinely conducted tests [152].
In conclusion, the real-world evidence summarized above is consistent with the findings of the phase 3 trial of VN [138]. The gene therapy was shown to provide an evident benefit in terms of light sensitivity in patients with RPE65-associated retinal dystrophy. The beneficial effect was observed regardless of the disease severity and manifested as early as 1 month postinjection. Additionally, the treatment produced some benefits in terms of visual field expansion and VA. The spectrum of adverse events was similar to that documented in the phase 3 trial [138], but some new perioperative complications were identified as well, most likely related to the lack of patients’ compliance with a 24-hour supine positioning after the treatment. Available real-world evidence suggests that the indications for VN treatment might be expanded on additional groups of patients, such as those with late-presentation RPE65-associated retinal dystrophy. Finally, published real-world data suggest that the VN treatment outcomes might benefit from some modifications to the surgical procedure and posttreatment monitoring protocol.
Conclusion
In this article, we reviewed the epidemiology, genetics and clinical features of RPE65-associated retinal dystrophy. Also, we discussed the eligibility, treatment procedures, clinical outcomes and safety of subretinal VN administration. From the clinical trials and real-world data, we can conclude that VN treatment significantly improves functional vision in RPE65-associated retinal dystrophy, a previously untreatable condition. Due to extremely low prevalence and phenotypic heterogeneity, it is often challenging to detect RPE65-associated retinal dystrophy. With the knowledge of phenotypic features of RPE65-associated retinal dystrophy and the availability of appropriate genetic testing, we may be able to identify more patients who are eligible for VN treatment.





 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print