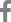Dear Editor,
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal degeneration that features degeneration of rod and cone photoreceptors [1]. The CFAP410 gene (previously known as C21orf2) encodes a ciliary protein that plays a role in cilia formation and/or maintenance and DNA damage repair [2]. Pathogenic variants in CFAP410 have been recently reported in a Japanese patient with autosomal recessive RP and skeletal anomalies [2]. Herein, we report the first case of CFAP410 (p.Tyr107His and p.Val111Met) compound heterozygous pathogenic variants in a Korean patient with RP and skeletal defects identified by whole-exome sequencing (WES).
This study was approved by the institutional review board at Samsung Medical Center (2014-09-052), and written informed consent was obtained from the patient’s parents. A 15-year-old female visited the retina clinic complaining of decreased visual acuity. Her best-corrected visual acuity in the right eye was 20 / 100 and left eye 20 / 70, and the refractive error (spherical equivalent) for the right eye and left eye was +1.5 and −2.5 diopters, respectively. On orthoptic evaluation, there was an alternating esotropia with small dissociated vertical deviation in both the eyes. There was horizontal, low-amplitude, and high-frequency jerk nystagmus.
Although she had no family history of visual impairment, genetic testing conducted later revealed an autosomal recessive inheritance pattern. She had a short stature (<1st percentile) and a history of surgical correction for pectus carinatum at the age of 3 years (Fig. 1A). Other bony abnormalities were not observed in the plain skeletal series. She had no other developmental problems. Fundus examination showed peripheral retinal degeneration with pigmentary deposits (Fig. 1B). Fundus autofluorescence showed patch hypoautofluorescent areas surrounded by ring-shaped hyper-autofluorescence (Fig. 1C). Optical coherence tomography showed retinal pigment epithelium atrophy, photoreceptor layer defects, and macular staphyloma (Fig. 1D). The visual field test showed severe visual field constriction (Fig. 1E). The full-field electroretinogram showed reduced amplitudes, especially in the cone responses, with relatively preserved rod responses (Fig. 1F).
For genetic diagnosis, genomic DNA of the patient and her parents were extracted from whole blood using QIAamp DNA mini kits (Qiagen, Valencia, CA, USA). Whole exome capture and sequencing were performed using the SureSelect XT Human All Exon v5 kit (Agilent Technologies, Santa Clara, CA, USA). The variants were filtered out with the following criteria: (1) variants with low variant allele fractions (<5%); (2) variants with minor allele frequency (>1%) in population databases, including Korean Reference Genome Databases and in-house knowledgebase of RP; (3) variants in genes not related to RP; and (4) variants in genes that do not match the inheritance pattern. WES revealed that the patient was compound heterozygous for two missense variants in the CFAP410 gene (GenBank accession no. NM_004928.2), which were c.319T>C:p.(Tyr107His), and c.331G>A:p.(Val111Met). Variants were confirmed by Sanger sequencing, with both parents being heterozygous carriers of each variant. These two variants have been previously reported as disease-causing variants [2]. According to the American College of Medical Genetics and Genomics recommendations on the interpretation of sequence variants, we classified these variants as likely pathogenic [2,3]. No other “pathogenic” or “likely pathogenic” variants or copy number changes were found in other RP-related genes. A previous in vitro study revealed the pathogenic effects of CFAP410 variants [2].
The possibility of axial spondylometaphyseal dysplasia was considered as accompanying RP and skeletal defects such as short stature and pectus carinatum were observed [4]. However, due to lack of characteristic abnormalities in other plain skeletal series, we could not classify this patient as having axial spondylometaphyseal dysplasia or Jeune syndrome. Khan et al. [5] reported that CFAP410 is mutated in recessive early onset retinal dystrophy with macular staphyloma without skeletal anomalies except for short stature. Our patient showed both macular staphyloma and skeletal anomalies. The skeletal phenotype in individuals with the CFAP410 mutation might be quite distinct.
In conclusion, we report a Korean patient with autosomal recessive RP and skeletal defects diagnosed by WES. Our patient had variants identical to those of a previous Japanese report with similar clinical features but showed differences including the presence of nystagmus, macular staphyloma, and more severe chest deformity (pectus carinatum) [2]. To the best of our knowledge, this is the first case of pathogenic variants in CFAP410 (p.Tyr107His and p.Val111Met) in the Korean population.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print