Glaucoma is a chronic, progressive optic neuropathy with a characteristic optic nerve head change and corresponding visual field (VF) defects. It is the leading cause of irreversible blindness worldwide. Primary open angle glaucoma (POAG) is the most common type of glaucoma and occurs in elderly individuals with an open angle without gonioscopic abnormalities. The overall prevalence of POAG ranges from 1.0% to 3.9%, depending on the race, age and definitions used [1,2,3].
Juvenile-onset open angle glaucoma (JOAG) is an uncommon subset of POAG characterized by an autosomal dominant pattern of inheritance [4,5]. It generally affects individuals during childhood or early adulthood but is distinct from congenital glaucoma that presents with buphthalmos, megalocornea, Haab's striae, and ocular or other systemic developmental anomalies. A recent population-based study reported that the incidence of JOAG was 0.38 per 100,000 residents between 4 and 20 years of age [6]. An epidemiological study from the Dallas Glaucoma Registry reported that JOAG comprised about 4% of the cases of childhood glaucoma when JOAG was defined as an idiopathic glaucoma arising in children older than three years of age [7]. JOAG is associated with myopia and severely elevated intraocular pressure (IOP) with large fluctuations [8,9].
The resulting visual impairment and blindness can significantly impair the patient's quality of life and limit daily living activities [10]. Patients with JOAG are diagnosed at an early age and therefore have a longer life expectancy than the typical glaucoma patient. Because of the rarity of the disease, there are little data on its clinical characteristics and prognosis. In this study, we demonstrated the clinical characteristics and evaluated the prognostic factors for the progression of VF defects in patients with JOAG.
Materials and Methods
This retrospective study was approved by the institutional review board of Samsung Medical Center and was carried out in accordance with the tenets of the Declaration of Helsinki. We reviewed the medical records of 125 eyes in 72 consecutive patients (15 eyes of 15 unilateral JOAG patients and 110 eyes of 57 bilateral JOAG patients) who had been diagnosed with JOAG at Samsung Medical Center between 1996 and 2014. JOAG was also diagnosed when patients met the following criteria: elevated IOP Ōēź24 mmHg by Goldmann applanation tonometry at the initial hospital visit, open angle configuration on gonioscopy, glaucomatous optic neuropathy (neural rim thinning, focal notching or a vertical cup-to-disc ratio >0.6) and/or glaucomatous VF defects, and patients between the ages of 10 and 40 years. The results of the medical history and comprehensive ophthalmologic examination, including slitlamp biomicroscopy, best-corrected visual acuity, refractive error, and central corneal thickness, were recorded. The IOP was measured on two separate occasions with a Goldmann applanation tonometer under topical anesthesia, and the average value of the two was determined and used. The baseline IOP was checked at the initial hospital visit, and the final IOP was calculated as the mean IOP measured during the last two hospital visits. An automated VF test was evaluated by the 30-2 program Swedish interactive threshold algorithm standard on the Humphrey 740 Visual Field Analyzer (HFA 30-2; Carl Zeiss Meditec, Dublin, CA, USA). A dilated stereoscopic examination of the fundus with optic disc and red-free fundus photography for identifying a glaucomatous optic nerve head, retinal nerve fiber layer defects and retinal pathology, was reviewed for all participants. The VF test, color optic disc photographs and red-free fundus photographs were acquired at an interval of one year. The evaluated medical history included the patient's family history of glaucoma, sex, follow-up duration, numbers of medications used for the management of IOP, steroid use, chief complaint at the initial visit, and age at the diagnosis of JOAG. A positive family history of glaucoma was defined as the presence of one or more relatives (first- or second-degree) of the patient who reportedly had been diagnosed with glaucoma by ophthalmologists. The JOAG patients were grouped according to the laterality of the disease and the VF progression.
Glaucomatous VF defects were defined as the presence of a cluster of three or more contiguous non-edge points on the pattern deviation probability plot with a probability less than 5%, with at least one of these having a probability less than 1%, which was confirmed on two consecutive tests. Unreliable test results were excluded if the fixation loss was more than 30%, or a false-positive or false-negative was greater than 33%. VFs were evaluated for progression using a modification of the criteria suggested by Anderson and Patella [11]. The first VF test was excluded to minimize the impact of the learning effect. At least four reliable VF datasets with a mean deviation better than -20.00 dB of the baseline VF test were required. Progression was defined when one of the following was present compared to the baseline values: a reproducible reduction in the sensitivity of at least 10 dB in a cluster of Ōēź2 contiguous locations and/or a deterioration of at least 5 dB in a cluster of Ōēź3 contiguous locations, at least one of which had deteriorated by Ōēź10 dB on two consecutive VF tests. The progression points were not the outermost points and did not cross the nasal meridian. Out of 125 eyes of 72 patients, 30 eyes had less than four reliable VF tests; 15 eyes had a mean deviation worse than -20.00 dB or threat to fixation on the baseline VF test. The remaining 80 eyes were included for the analysis of the VF progression.
Exclusion criteria were as follows: (1) evidence of secondary causes of elevated IOP; (2) a history of intraocular surgery, including laser treatment and refractive surgery; (3) pigmentation of the angle greater than grade 3 or peripheral anterior synechia; (4) conditions other than glaucoma affecting the VF; (5) presence of any other retinal or neurological pathology; and (6) no light perception of visual acuity.
Statistical analyses were performed using SAS ver. 9.4 (SAS Institute, Cary, NC, USA) and R 3.0.3 (R Foundation for Statistical Computing, Vienna, Austria; http://www.R-project.org/). We used the independent t-test and chi-square test to compare the means of data within the JOAG subgroups. For a comparison between bilateral and unilateral JOAG patients, eyes diagnosed with JOAG were included in our statistical analyses. In bilateral JOAG patients, the eye presenting with a higher IOP at the initial visit among both eyes was used. All eyes were diagnosed with JOAG, and both eyes in the case of bilateral JOAG patients were included in the statistical analysis to compare the groups when they were divided according to the VF progression. To identify the clinical parameters associated with VF progression, each variable was first tested in a univariable model using logistic regression analysis. Those with a p-value less than 0.10 were then entered in a multivariable model. The correlations of the outcomes of both eyes in one patient were adequately adjusted. To do this, a generalized estimating equations method was adopted in our analyses. A p-value less than 0.05 was considered statistically significant.
Results
We enrolled 125 eyes of 72 patients with JOAG, of which there were 46 males and 26 females. Four eyes presenting with no light perception were excluded from this study because they are typically not treated when the patients do not complain of pain. At the initial hospital visit, 15 patients (21%) were diagnosed with unilateral JOAG. During the follow-up, nine fellow normal eyes were diagnosed with JOAG, and three of nine eyes demonstrated a glaucomatous VF defect. Patient characteristics and comparisons between bilateral and unilateral JOAG patients are shown in Table 1. Among the 72 JOAG patients, the mean age at diagnosis was 26.8 ┬▒ 7.3 years old, and the mean follow-up duration was 94.4 ┬▒ 50.5 months. Eighteen patients (28%) had a known family history of glaucoma, while 46 (72%) did not. Eight patients (11%) were not sure of their history of glaucoma. The mean refractive error corrected for spherical equivalence was -4.99 ┬▒ 4.01 diopters. Of these patients, 57 (79%) had glaucoma in both eyes, and they were defined as the bilateral group. The age at diagnosis was significantly older in the bilateral group (27.8 ┬▒ 7.4 years) than in the unilateral group (23.3 ┬▒ 5.7 years, p = 0.039), and the bilateral group (32%) more commonly showed a family history of glaucoma than the unilateral group (9%, p = 0.035). Other parameters were not significantly different between the bilateral and unilateral groups. Fifty-six eyes (44.8%) were successfully controlled with medical treatment, and the mean IOP at the last visit was 18.35 ┬▒ 3.50 mmHg with an average of 2.43 ┬▒ 1.13 eye drops. Sixty-nine eyes (55.2%) underwent surgical treatment, and the mean IOP at their last visit was 14.17 ┬▒ 4.72 mmHg with an average of 1.14 ┬▒ 1.50 eye drops.
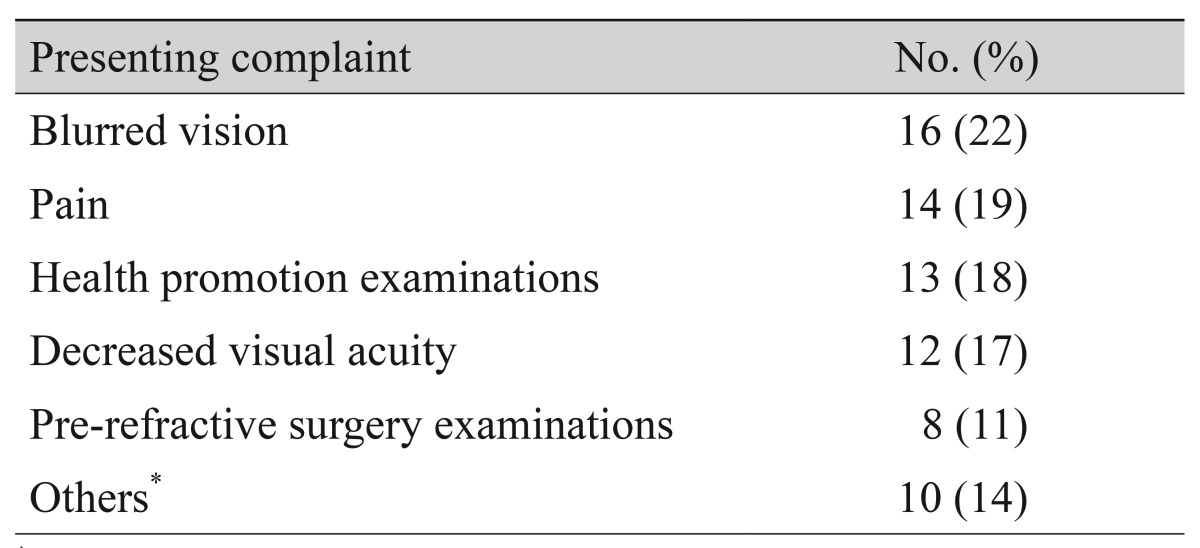
The chief complaints of patients with JOAG at their initial hospital visit are shown in Table 2. Blurred vision was the most common manifestation in 16 (22%), and 14 reported pain (19%). Twenty-one patients (29%) were diagnosed with glaucoma by health promotion and pre-refractive surgery examinations. Others included conjunctival injection, tearing, irritation, and foreign body sensation.
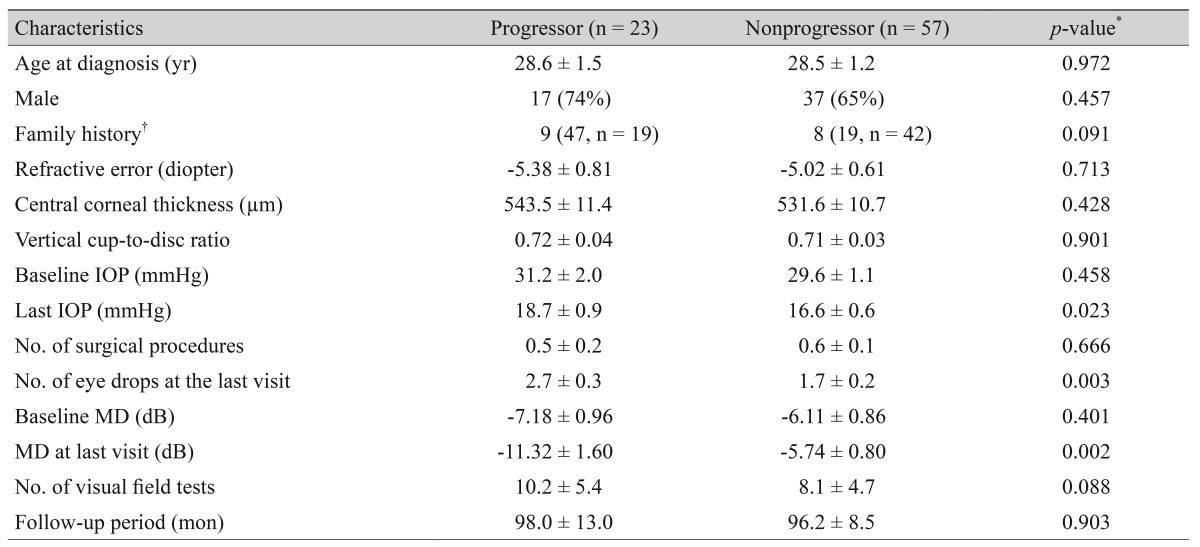
Table 3 shows a comparison of the clinical characteristics between 23 progressors and 57 nonprogressors in 80 eyes with JOAG. The progressor group (47%) represented a higher proportion of the family history than the nonprogressor group (19%, p = 0.091), but the difference was not significant. The last IOP was significantly higher in the progressor group (18.7 ┬▒ 0.9 mmHg) than in the nonprogressor group (16.6 ┬▒ 0.6 mmHg, p = 0.023), despite the significantly higher number of IOP-lowering eye drops being used at the last visit in the progressor group (2.7 ┬▒ 0.3) vs. the nonprogressor group (1.7 ┬▒ 0.2, p = 0.003).
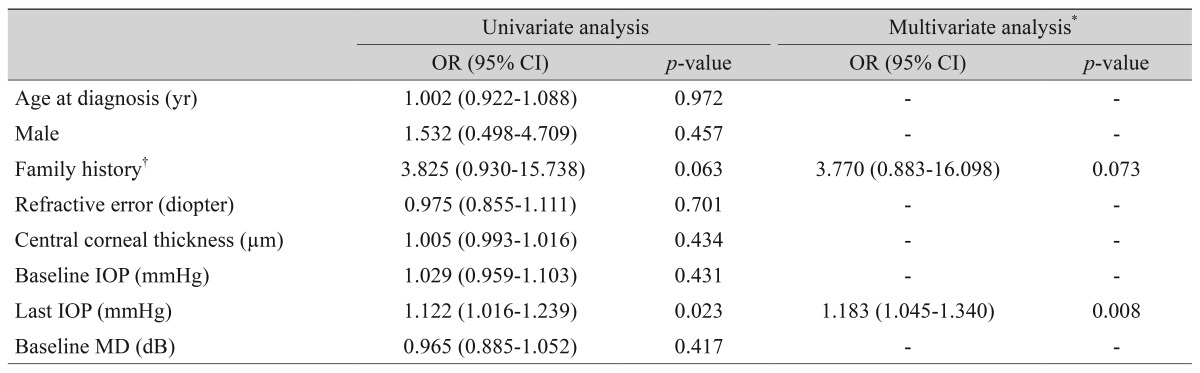
The univariate logistic regression analyses are shown in Table 4. The last IOP showed significant associations with the VF progression (odds ratio [OR], 1.122; p = 0.023). Variables with p-values of less than 0.10 were included in the multivariate analysis to assess their joint effects on the VF progression. Using multivariate analyses, the VF progressions were associated with the final IOP (OR, 1.183; p = 0.008).
Discussion
In this study, we demonstrated that JOAG patients had a male preponderance, myopic refractive state and severe elevation of IOP; these findings were similar to those of previous reports [8,12,13,14]. Bilateral JOAG patients were older and made up a higher proportion of those with a family history of glaucoma than unilateral JOAG patients. It is probable that unilateral JOAG patients were diagnosed earlier than bilateral JOAG patients, because 64% of the normal fellow eyes of unilateral JOAG patients at the initial hospital visit were also diagnosed with JOAG during the follow-up in this study. In cases of normal tension glaucoma, one-fourth of patients developed VF loss in the fellow eyes that had an initially normal VF, and the estimate of the median time to VF loss onset was 95.1 months [15]. In this study, nine of 14 patients (64%) with unilateral JOAG developed JOAG in their fellow eyes, and the estimate of the median time to diagnosis was 12.3 months. Because the patients with unilateral JOAG had higher chance of developing JOAG in their fellow normal eyes, more caution is needed during follow-up to identify these patients so they can receive proper treatment.
Symptoms associated with visual acuity, including blurred vision and decreased visual acuity, were the major manifestation. However, one-third of the patients visited the clinics without definite symptoms and was diagnosed with JOAG by chance. Visual impairment and vision-related quality of life in working-age adults was related with impaired vision as well as with adverse social outcomes [10]. Because JOAG patients have a long life expectancy, the periodic IOP measurements conducted during the school's physical examinations are important despite their rarity.
Our study showed that VF progression was associated with a higher IOP at the last hospital visit. A family history of glaucoma was weakly associated with a higher proportion of VF progression despite there being no significant differences in the baseline and last IOP, number of surgical procedure and eye drops and the baseline VF defect. In the case of POAG in older patients, an evidence-based review showed that there was no significant relation between the patient's family history of glaucoma and VF progression [16]. In contrast, Wu et al. [17] observed that patients with familial POAG had a greater disease severity and an earlier onset age at diagnosis compared to patients with sporadic disease. Previous studies have reported that the relation between a family history of glaucoma and the progression or severity of the VF defect was controversial in POAG patients. However, there have been no such reports in JOAG patients. Souzeau et al. [18] found that the prevalence of myocilin mutations in glaucoma cases with severe VF loss was significantly greater than in nonadvanced glaucoma patients. In addition, the prevalence of a myocilin mutation of POAG patients ranged from 1.4% to 4.3%, worldwide [19,20,21], while 12.5% to 36% of cases of JOAG might have been linked to myocilin mutation [22,23]. Therefore, it is possible that a family history in JOAG patients might be associated with VF progression.
This study had several limitations. First, because of its retrospective design, there were possibly confounding factors and selection bias. In the event of poorly managed patients, drop-out during follow-up may have occurred. However, there was no significant difference in the clinical backgrounds, including the age, sex, baseline IOP, duration of follow-up, and the severity of VF defect at initial visit between the VF progression and the nonprogression groups. The second limitation was the relatively small number of total patients due to its rarity, which made it difficult to detect differences among groups.
In conclusion, we confirmed that JOAG patients presented with a male preponderance, a myopic refractive state and severe elevation of IOP. Periodic eye examinations are needed because a considerable number of JOAG patients have no definite symptoms. Our data showed that the IOP at the last hospital visit was higher in patients were experiencing a progression of VF defects. These findings indicated that JOAG patients may require more careful management of IOP. For a definite conclusion, further large-scale prospective studies would be needed.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print