Dear Editor,
X-linked retinitis pigmentosa (XLRP) associated with pathogenic variants in the retinitis pigmentosa GTPase regulator (RPGR) gene is a severe form of RP featuring early-onset nyctalopia and rapid deterioration of visual acuity in male patients, frequently leading to legal blindness within the fourth decade of life [1]. RPGR-related XLRP accounts for approximately 70% of XLRP cases [2] and has been reported to manifest mostly in males who carry the pathogenic allele in a hemizygous form.
Interestingly, female carriers of pathogenic RPGR variant have been reported to exhibit variable phenotypes of clinical severity ranging from being asymptomatic to having significant visual loss [3]. Herein, we report two cases of female carriers with RPGR mutation showing severe clinical manifestations comparable to male patients. Written informed consents for publication of the research details and clinical images were obtained from patients.
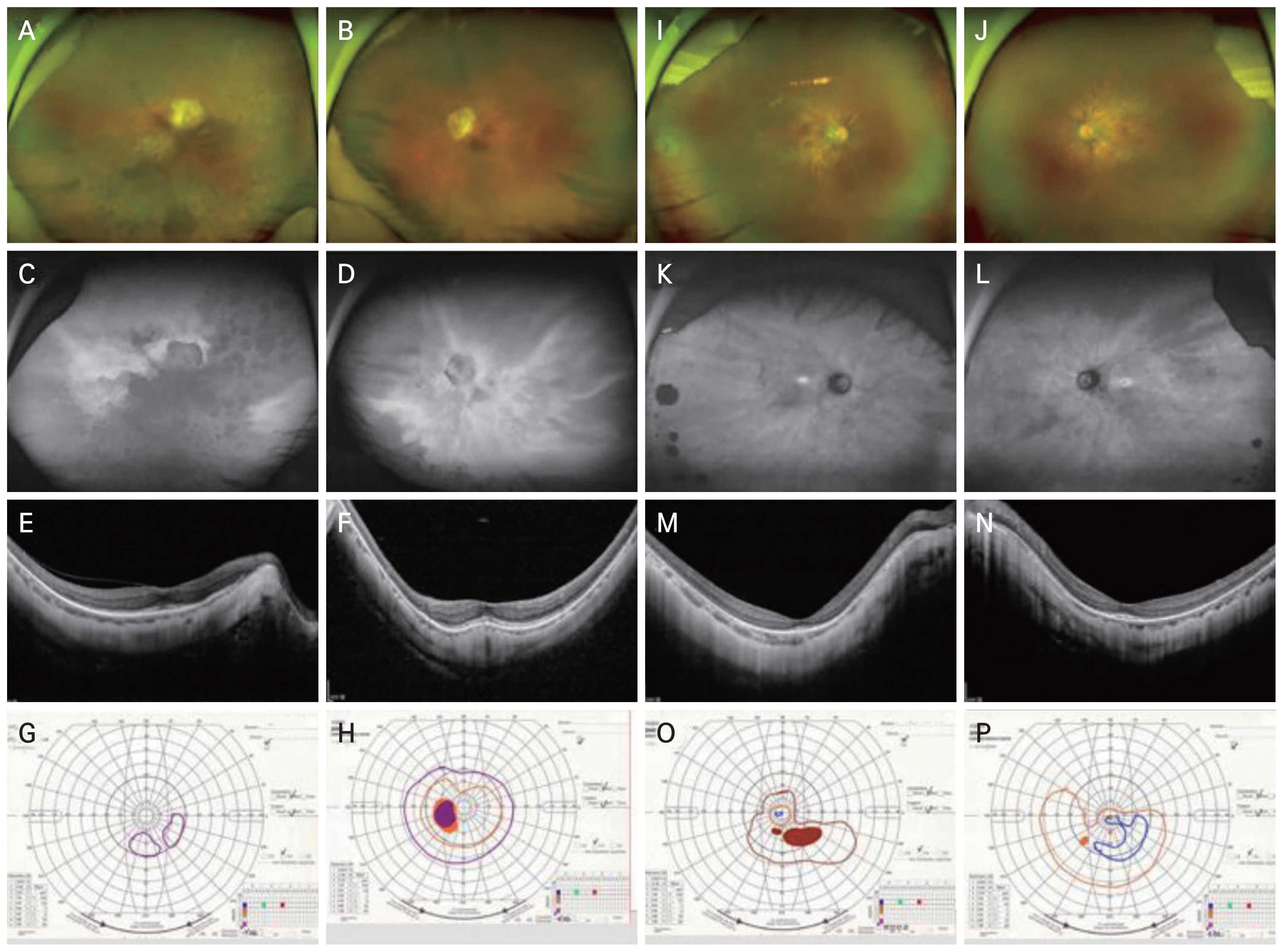
Case 1 is a 74-year-old Korean woman presented to the inherited retinal degeneration clinic with decreased visual acuity and blurry vision in both eyes for 1 year. From early childhood, she suffered from night blindness. Her only son, who also had night blindness and visual field constriction, had been diagnosed with RP at the age of 20 years. On presentation, her decimal best-corrected visual acuity was 0.03 in the right eye and 0.10 in the left eye. Fundus examination showed extensive degeneration with bony spicules in both eyes (Fig. 1A, 1B). Optical coherence tomography confirmed outer retinal loss, and f undus autofluorescence revealed diffuse peripheral hypofluorescent spots (Fig. 1C-1F). International Society for Clinical Electrophysiology of Vision (ISCEV)-standard full-field electroretinogram showed nearly flat rod and cone response in both eyes. Severe visual field constriction in the right eye and constriction of the visual field along with a widening of the physiological blind spot in the left eye, were observed (Fig. 1G, 1H). The asymmetry was due to the central media opacity and more diffuse pigmentary changes in the right eye. Next-generation sequencing-based panel tests targeting RP discovered a heterozygous RPGR gene mutation (reference sequences NM_001034853.1, c.491G>A p.(Trp164Ter)), classified as a “likely pathogenic” mutation according to the American College of Medical Genetics and Genomics (ACMG/AMP) guidelines.
Case 2 is a 38-year-old Korean woman was referred to the retina clinic due to deteriorating night vision and visual acuity for 1 year. In the daylight, she had difficulty focusing on written letters, and the letters seemed partially erased. In the dark, her visual field was obscured by dark gray, except for the inferonasal part of the right eye and the central field of the left eye. Her mother also had high myopia and mild night blindness. Her decimal best-corrected visual acuity was 0.3 in the right eye and 0.5 in the left eye. Fundus revealed retinal vessel attenuation (Fig. 1I, 1J). Parafoveal ring-shaped abnormal autofluorescence and characteristic tapetal-like reflex were seen in autofluorescence imaging (Fig. 1K, 1L). Corresponding to the hyperautofluorescent spots, parafoveal loss of the outer retinal layers was found on optical coherence tomography (Fig. 1M, 1N). Kinetic perimetry showed peripheral visual field loss in both eyes (Fig. 1O, 1P). According to the next-generation sequencing panel testing, a heterozygous RPGR gene mutation (reference sequencing NM_001034853.1, c.1414+1G>T) was detected and classified as a “likely pathogenic” mutation per ACMG/AMP guidelines.
RP is the most common form of inherited retinal degenerations, inherited in diverse patterns, including XLRP. XLRP tends to manifest a more severe clinical course compared to other forms. It is typical for heterozygous females to remain asymptomatic, while only hemizygous males are affected. However, it has long been appreciated that female carriers may also exhibit a spectrum of clinical presentations, from very mild to even severe. This phenomenon results from variable degrees of the X-chromosome inactivation ratio. X-chromosome inactivation serves as a gene dosage compensation mechanism in mammals to achieve equilibrium in the expression of X-linked gene across genders [4].
This report represents the first documented cases of Korean female carriers with severe phenotypes associated with RPGR-related XLRP. Due to the phenotypic diversity of female carrier, XLRP pedigrees with affected females but no male-to-male transmission may be misinterpreted as autosomal dominant [5]. This report will help identify the disease entity and overcome the diagnostic challenge of XLRP. RPGR gene therapy is currently under development, and should a therapeutic intervention be developed in the future, it could be considered for inclusion in the treatment for female patients who manifest a phenotype of comparable severity to their male counterparts.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print